اختلالات متابولیک
اختلالات متابولیک
اختلالات متابولیک
اختلالات متابولیک
اختلالات اسیدآمینه ها
اسیدهای آمینه انواعی از مولکول های زیستی هستند که به عنوان اجزا ساختاری پروتئین ها عمل می کنند. در مجموع بیست نوع اسید آمینه مختلف در ساختار پروتئین ها دیده می شود و تعدادی اسید آمینه دیگر نیز در بدن موجود هستند که در واقع حد واسط های متابولیکی سایر اسید های آمینه بوده و نقصان در سنتز و متابولیسم آنها منجر به بروز برخی انواع بیماری می شود. در راس تمایم بیماری های متابولیک مرتبط با اسید های آمینه بیمای فنیل کتونوری قرار دارد. این بیماری که در سال 1953 توسط Jervis معرفی شد اولین بیمای است که در آن مشخص گردید عامل بروز بیماری نقص در یک آنزیم می باشد. در مبحث مربوط به فنیل کتونوری به صورت مفصل در خصوص آن بحث شده است.
اسید آمینه چیست؟
اسید های آمینه مولکول های زیستی با ساختار مشخصی هستند که در تصویر زیر می توانید این ساختار را مشاهده کنید. در هر اسید آمینه یک مولکول کربن مرکزی وجود دارد که به آن همواره سه گروه جانبی ثابت و یک گروه جانبی متغییر متصل شده است. گروه های ثابت شامل یک مولکول هیدروژن، یک گروه کربوکسیل (COOH) و یک گروه آمین (NH2) می باشد که این سه گروه در تمامی اسید های آمینه یکسان است، خصوصیت هر اسید آمینه توسط گروه چهارم تعیین می شود که در تصویر اول با حرف R نمایش داده شده و در تصویر دوم انواع گروه های را می توانید ببینید.
.
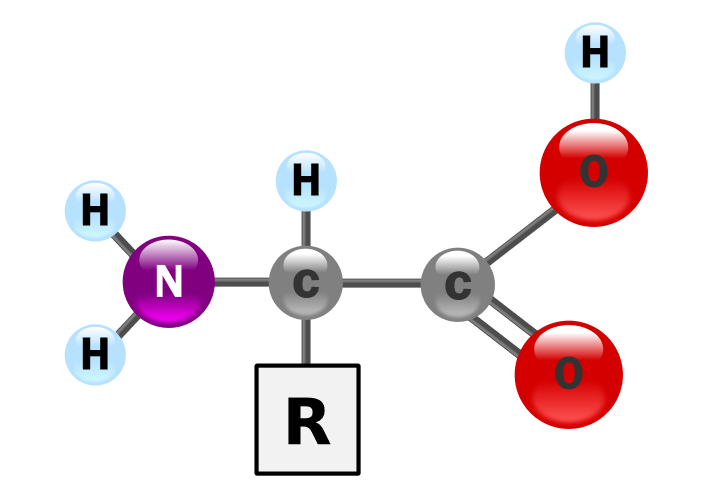
تصویر 2- انواع گروه های جانبی در اسید های آمینه
هر یک از گروه های جانبی می توانند دارای بار الکتریکی یا فاقد آن باشند، می توانند قطبی یا غیر قطبی باشند و یا آنهایی که بار دارند می توانند بار مثبت یا منفی داشته باشند. بر این اساس بیست نوع اسید آمینه با هویت و خصوصیات متفاوتی شکل می گیرد که زمانی که آنها در کنار هم در ساختار پروتیین ها قرار می گیرند بر اساس نحوه چینش شان در کنار یکدیگر خصوصیات متفاوتی را به پروتئین ها می دهند و عملا تعداد بی نهایت ترکیب می تواند با این بیست نوع اسید آمینه ساخته شود.
انواع اختلالات متابولیسم اسید های آمینه
به طور خلاصه بیماری های متابولیسم اسید های آمینه را می توان به چندین زیر گروه مختلف به شرح زیر تقسیم بندی نمود:
اختلالات چرخه اوره و هایپر آمونمی وراثتی
چرخه اوره مسیر متابولیسمی 5 مرحله ای است که در سلولهای کبدی اتفاق می افتد و طی آن نیتروژن زاید حاصل از گروه های آمین اسید های آمینه در فرآیند تجزیه پروتئین ها حذف و دفع می گردد. به طور خلاصه در طی این چرخه دو مولکول آمونیاک و یک مولکول بی کربنات به یک مولکول اوره تبدیل می شود (اوره شکل بی خطر نیتروژن مازاد در بدن می باشد که به راحتی از طریق ادرار دفع می شود). نقص در هر یک از آنزیم های چرخه اوره باعث عدم تحمل پروتئین به دلیل افزایش آمونیاک (هایپرآمونمیا) می شود. افزایش آمونیاک برای سیستم اعصاب مرکزی سمی است و می تواند منجر به کما و یا حتی مرگ شود. بیماری های مختلفی در این گروه دیده می شود که به جز نقص اورنیتین ترانس کربامیلاز که حالت توارث وابسته به جنس دارد سایر آنها از الگوی توارث اتوزومی مغلوب پیروی می کنند (برای کسب اطلاع در مورد الگوی اتوزومی مغلوب و یا وابسته به جنس کلیک کنید). بیماری های این گروه شامل موارد زیر می باشد:
- سیترولینمی
بیماری سیترولینمی، یکی از اختلالات سیکل اوره است. این بیماری به علت عدم حضور و یا فعالیت نادرست آنزیم آرژنینو سوکسینیک اسید سینتاز به وجود می آید. وظیفه این آنزیم، هضم برخی اسیدهای آمینه و دفع آمونیاک بدن است. وقتی که آنزیم آرژنینو سوکسینیک اسید سینتاز به درستی فعالیت نکند، سطح اسید آمینه سیترولین، آمونیاک و دیگر مواد سمی در بدن افزایش می یابد. افزایش سطح این مواد در خون، می تواند باعث ایجاد آسیبهای مغزی و حتی مرگ شود.
اگر بیماری سیترولینمی درمان نشود، چه مشکلاتی به وجود می آید؟
آمونیاک ماده ای سمی است به همین دلیل قبل از دفع شدن به اوره تبدیل می شود. اوره ماده ای غیر سمی بوده و به راحتی از طریق ادرار دفع می گردد. اگر آمونیاک به اوره تبدیل نشود، غلظت آن در خون افزایش می یابد. افزایش سطح آمونیاک خون برای مدت طولانی، آسیبهای مغزی را به وجود خواهد آورد. علایم بیماری سیترولینمی و زمان بروز این علایم، در افراد مختلف، متفاوت است. سیترولینمی دو شکل مختلف دارد که فرم متداول آن سیترولینمی کلاسیک نامیده می شود. زمان بروز علایم در سیترولینمی نوع کلاسیک، دوران نوزادی است. زمان بروز علایم در فرم خفیف تر این بیماری، معمولا کمی دیرتر است. بیماری سیترولینمی بزرگسالان نیز وجود دارد، که بسیار نادر است و در کشور ژاپن شیوع بیشتری دارد.
سیترولینمی کلاسیک
نوزادان مبتلا به سیترولینمی کلاسیک در بدو تولد به نظر سالم می رسند، اما پس از چند روز، سطح آمونیاک خون آنها افزایش یافته و علایم بیماری به سرعت در آنها بروز می کند. مهمترین علایم افزایش آمونیاک خون عبارتند از:
کاهش اشتها
احساس خواب آلودگی مفرط
حساسیت زیاد، کج خلقی و تحریک پذیری
استفراغ
اگر افزایش سطح آمونیاک خون درمان نشود، می تواند علایم زیر را ایجاد کند:
ضعف ماهیچه ها
شلی یا سفتی غیر طبیعی ماهیچه ها
مشکلات تنفسی
احساس سرما
تشنج
تورم مغز
کوما که در برخی مواقع به مرگ بیمار منجر می شود
دیگر تاثیرات بیماری سیترولینمی در برخی افراد، عبارتند از:
رشد کم
بزرگی کبد
مشکلات یادگیری و یا عقب ماندگی ذهنی
اغلب کودکان درمان نشده، در اولین هفته های بعد از تولد فوت می کنند.
سیترولینمی خفیف
علایم سیترولینمی نوع خفیف معمولا کمی دیرتر از فرم کلاسیک بروز می کند. علایم این بیماری، در کودکان درمان نشده عبارت است از:
رشد کم، مشکلات یادگیری یا عقب ماندگی ذهنی
موهای خشک و شکننده، پرهیز از گوشت و دیگر غذاهای پروتئینی
بیش فعالی، تشنج
مشکلات رفتاری، حملات ناگهانی، افزایش آمونیاک خون
افزایش آمونیاک خون، معمولا در موارد زیر ایجاد می گردد:
بعد از یک دوره گرسنگی
در هنگام عفونت و بیماری
بعد از دریافت مقدار زیادی پروتئین
برخی از علایم افزایش آمونیاک خون در کودکان عبارت است از:
کاهش اشتها احساس خواب آلودگی و ضعف مفرط
سردرد های شدید اختلال در تکلم
استفراغ عدم تعادل
اگر افزایش آمونیاک خون درمان نشود، می تواند علایم زیر را ایجاد کند:
مشکلات تنفسی
تورم مغز
تشنج
کوما که گاهی به مرگ بیمار منتهی می شود
در تعدادی از بیماران، هیچگاه علایم بیماری بروز نمی کند و این افراد هنگامی شناسایی می شوند که یکی از اعضای خانواده آنها که مبتلا به این بیماری است، شناسایی شود.
درمان سیترولینمی چیست؟
پزشک کودک شما با یک پزشک متخصص بیماریهای متابولیک و یک متخصص تغذیه همکاری خواهد کرد تا کودک شما را درمان کند. برای جلوگیری از عقب ماندگی ذهنی و مشکلات جسمی درمان فوری مورد نیاز است. شما به محض آگاهی از بیماری فرزندتان، باید معالجه را آغاز کنید. در ادامه روش درمانی که به طور معمول برای بیماران سیترولینمی پیشنهاد می شود، ذکر شده است :
رژیم غذایی با حداقل مقدار پروتئین، غذاهای طبی و شیرخشکهای مخصوص
اغلب کودکان مبتلا به سیترولینمی به یک رژیم غذایی خاص که شامل غذاهای کم پروتئین است، نیازمندند. غذاهای طبی خاص و شیر خشک های مخصوص بخشی از این رژیم غذایی هستند. متخصص تغذیه به تدریج رژیم غذایی مناسب کودک شما را، طراحی خواهد کرد. این برنامه غذایی باید در طول زندگی کودک شما رعایت شود.
رژیم غذایی با حداقل مقدار پروتئین
موثرترین درمان در بیماران مبتلا به سیترولینمی، رعایت رژیم غذایی کم پروتئین است. غذاهایی که حاوی پروتئین بالا بوده باید از رژیم حذف شده و یا مقدار آنها شدیدا محدود شوند عبارتند از: شیر و ترکیبات لبنی، گوشت و مرغ، ماهی، تخم مرغ، سبزیجات خشک شده، باقلا خشک شده و مغزها (بادام، پسته، گردو و غیره). دریافت مقدار زیادی از این مواد خوراکی، باعث افزایش سطح آمونیاک خون می شود. بسیاری از میوه ها و سبزیجات مقدار کمی پروتئین دارند و می توانند به مقدار معینی استفاده شوند. تمام پروتئین را از رژیم غذایی کودک حذف نکنید. کودکان مبتلا به سیترولینمی به مقداری پروتئین برای رشد مناسب نیازمندند. هرگونه تغییر در رژیم غذایی باید با اجازه متخصص تغذیه باشد.
غذاهای طبی و شیر خشکهای مخصوص
غذاهای طبی همچون آرد های کم پروتئین، ماکارونی و برنج ساخته شده اند که مناسب افراد مبتلا به اختلالات اسیدهای آمینه می باشند. متخصص تغذیه به شما خواهد گفت که چگونه می توانید از این غذاها در برنامه غذایی کودکتان استفاده کنید. علاوه بر رژیم غذایی کم پروتئین، ممکن است به کودک شما شیر خشک های خاص نیز تجویز شود. این شیر خشک ها، حاوی مقادیر مناسب پروتئین و دیگر مواد غذایی لازم برای رشد کودک است. پزشکان به شما خواهند گفت که کدام شیر خشک و چه مقدار از آن برای کودک شما مناسب است. برنامه غذایی فرزند شما به چندین پارامتر وابسته است همانند: سن کودک، وزن کودک، حال عمومی کودک و روند درمان کودک.
تجویز دارو
مصرف برخی داروها، به رهایی بدن از آمونیاک کمک می کند. این داروها از افزایش سطح آمونیاک خون جلوگیری می کنند. پزشک معالج به شما خواهد گفت که کودک شما به این داروها نیاز دارد یا خیر. کودکانی که علایم افزایش آمونیاک را دارند، بهتر است در بیمارستان بستری شوند. در بیمارستان معمولا داروهای کاهش دهنده آمونیاک خون، به صورت داخل وریدی به این بیماران داده می شود. گاهی برای کاهش آمونیاک خون از روش دیالیز نیز استفاده می شود. برای کودکان مبتلا به سیترولینمی استفاده از آرژنین متداول می باشد. آرژنین که یک اسید آمینه است، می تواند به رهایی بدن، از آمونیاک کمک کند. پزشک به شما خواهد گفت که کودک شما به آن نیازمند است، یا خیر. به یاد داشته باشید که هیچ دارویی را بدون مشورت با پزشک استفاده نکنید.
انجام تستهای منظم آزمایشگاهی
کودک شما به انجام تستهای مکرر خون به منظور کنترل سطح اسیدهای آمینه و آمونیاک نیاز خواهد داشت. نتیجه این تستهای آزمایشگاهی، رژیم غذایی و داروهای کودک شما را تعیین خواهند کرد. اگر کودک شما، علایم هر نوع بیماری را دارد فورا با پزشک تماس بگیرید. در برخی از کودکان مبتلا به سیترولینمی حتی یک بیماری مختصر، می تواند منجر به افزایش آمونیاک خون شود. برای پیشگیری از هر مشکلی، به محض اینکه کودکتان علایم زیر را داشت فورا با پزشک تماس بگیرید:
کاهش اشتها تب
احساس ضعف و فقدان انرژی عفونت و بیماری
خواب آلودگی تغییرات رفتاری و شخصیتی
استفراغ اختلال در راه رفتن و عدم تعادل
کودکانی که علایم افزایش آمونیاک خون را دارند، بهتر است که در بیمارستان بستری شوند. در این باره با پزشک معالج مشورت کنید.
وقتی سیترولینمی درمان می شود چه اتفاقی رخ می دهد؟
اغلب کودکانی که بعد از تولد سریعا تحت درمان قرار گرفته اند، اکنون زندگی عادی همراه با رشد و نمو و هوش طبیعی دارند. درمان فوری احتمال افزایش آمونیاک خون را کاهش می دهد. در برخی از بیماران، علیرغم درمان، باز هم حملات ناگهانی افزایش آمونیاک خون دیده می شود. در این کودکان آسیبهای مغزی، مشکلات یادگیری، عقب ماندگی ذهنی و تشنج مشاهده می شود.
چه عواملی باعث عدم حضور و یا فعالیت نادرست آنزیم می شود؟
کنترل ساخت آنزیم ها در بدن به عهده ژنها است. افراد مبتلا به سیترولینمی یک جفت ژن معیوب دارند که به درستی عمل نمی کنند. به خاطر این تغییر ایجاد شده در ژنها، یا آنزیم آرژنینو سوکسینیک اسید سینتاز ساخته نمی شود و یا آنزیم ساخته شده به درستی عمل نمی کند.
وراثت سیترولینمی چگونه است؟
این بیماری در دختران و پسران به یک نسبت مشاهده می شود. هر فرد دارای دو ژن است که دستور ساخت آنزیم را میدهد. در کودکان مبتلا به سیترولینمی هیچ یک از این دو ژن به درستی عمل نمی کنند. این کودکان هر یک از ژنهای معیوب را از یکی از والدین خود به ارث برده اند. به این نوع توارث، وراثت اتوزومال مغلوب می گویند. در والدین این کودکان، به ندرت اختلالی مشاهده می شود. اما هر یک از والدین یک تک ژن معیوب برای بیماری سیترولینمی دارد. این والدین ناقل نامیده می شوند. ناقلین علامت بیماری را بروز نمی دهند زیرا دیگر ژن آنها سالم است. وقتی هر دو والدین ناقل باشند، ۲۵% احتمال تولد کودک کاملا سالم،۵۰% احتمال تولد کودک ناقل همچون والدین و ۲۵% احتمال تولد کودک مبتلا به سیترولینمی در هر بارداری وجود دارد. مراکز مشاوره ژنتیک و مشاوران ژنتیک برای پاسخگویی به سوالات خانواده ها در دسترس می باشند. این مشاوران نحوه توارث بیماری را توضیح خواهند داد و همچنین راهنمای خانواده ها برای بارداری بعدی و مشاور آنها برای انجام تستهای تشخیصی برای دیگر اعضای خانواده خواهند بود.
آیا آزمایش ژنتیک خاصی در دسترس می باشد؟
امکان انجام تست ژنتیک وجود دارد. آزمایش ژنتیک که آزمایش DNA نیز نامیده می شود، تغییرات ایجاد شده در ژنها را بررسی می کند. در صورت هرگونه ابهام درباره آزمایش ژنتیک با پزشک متخصص بیماریهای متابولیک و یا با مشاور ژنتیک خود مشورت کنید. ممکن است، انجام آزمایش ژنتیک برای تشخیص بیماری ضروری نباشد، اما اگر امکان آن وجود داشته باشد، می تواند برای تشخیص قبل از تولد و یا تشخیص ناقلین مفید باشد.
آیا آزمایشهای تاییدی وجود دارند؟
سیترولینمی به وسیله تستهای خاصی که بر روی نمونه خون و ادرار و پوست انجام می شوند، تایید می گردد. درباره این تستها با مشاور ژنتیک و یا پزشک متخصص بیماریهای متابولیک مشورت کنید.
آیا شما می توانید در طول بارداری آزمایش انجام دهید؟
اگر یکی از فرزندان شما مبتلا به سیترولینمی است، شما میتوانید آزمایش DNA را در طول بارداری بعدی انجام دهید. این آزمایش به شما خواهد گفت که، کودک بعدی شما مبتلا به این بیماری هست یا خیر. نمونه مورد نیاز برای انجام این تست از آمینوسنتز و یا CVS به دست می آید. اگر نتیجه آزمایش DNA قانع کننده نبود، تستهای آنزیمی دیگری نیز قابل انجام هستند. نمونه مورد نیاز برای این تستها از آمینوسنتز و یا CVS به دست می آید. والدین میتوانند تصمیم بگیرند که، تستهای آزمایشگاهی را در طول بارداری بر روی جنین انجام دهند و یا بعد از تولد کودک، تستها را روی نوزاد انجام دهند. مشاوران درباره این که کدام روش بهتر است به والدین توضیح خواهند داد.
آیا امکان دارد که، دیگر اعضای خانواده مبتلا به سیترولینمی و یا ناقل آن باشند؟
امکان اینکه افراد دیگر خانواده، به این بیماری مبتلا باشند وجود دارد، هرچند علایم بیماری در آنها مشاهده نشود. جستجوی بیماری در دیگر اعضای خانواده بسیار مفید خواهد بود، زیرا از مشکلات جسمی پیشگیری خواهد کرد. از مشاور ژنتیک و یا پزشک متخصص سوال کنید که آیا دیگر اعضای خانواده باید تحت آزمایشهای تشخیصی قرار گیرند یا خیر. برادران و خواهران دیگر بیمار که سیترولینمی ندارند ممکن است همانند والدین خود ناقل باشند. در ناقلین هیچ اختلالی مشاهده نمی شود. تستهای تشخیص ناقلین به جز در موارد خاص فقط در افراد بالای ۱۸ سال قابل انجام است. برادران و خواهران هریک از والدین نیز ۵۰% احتمال دارد که ناقل این بیماری باشند. اطلاع از ناقل بودن از این جهت که آنها نیز ممکن است بچه های مبتلا به این بیماری داشته باشند، حایز اهمیت است. هنگامی که هر دو والدین می دانند که آنها ناقل بیماری هستند و یا زمانی که یک کودک مبتلا به بیماری در خانواده وجود دارد، دیگر کودکانی که در این خانواده ها متولد می شوند باید علاوه بر آزمایشهای غربالگری تحت آزمایشهای تشخیصی دقیقتر، برای سیترولینمی قرار گیرند.
آیا امکان انجام آزمایش در دیگر اعضای خانواده وجود دارد؟
تستهای تشخیصی:
امکان انجام تستهای تشخیصی در دیگر افراد خانواده وجود دارد. نمونه مورد نیاز برای این تستها، نمونه خون، ادرار و پوست است.
تستهای تشخیص ناقلین:
اگر یکی از افراد خانواده مبتلا به سیترولینمی است، امکان انجام تست DNA در دیگر اعضای خانواده وجود دارد. اگر نتیجه این تست قانع کننده نبود، شما می توانید دیگر متدهای تشخیص ناقلین را انجام دهید. درباره این تستها با پزشک خود مشورت کنید.
شیوع بیماری سیترولینمی چقدر است؟
شیوع این بیماری در ایالت متحده آمریکا در حدود ۱ مورد در هر۵۷۰۰۰ تولد است. این بیماری در تمامی نژادهای قومی سرتاسر جهان وجود دارد. شیوع بیشتر این بیماری در نژاد، قبیله، موقعیت جغرافیایی و یا کشور خاصی گزارش نشده است، هرچند سیترولینمی نوع بالغین در کشور ژاپن شیوع بیشتری دارد. پیش بینی می شود، این بیماری در کشورهایی که ازدواجهای فامیلی رواج بیشتری دارد (همانند کشور ایران) شایعتر باشد.
آیا این بیماری نامهای دیگری نیز دارد؟
این بیماری گاهی با نامهای زیر خوانده می شود:
سیترولینمی نوع ۱ (فرم کلاسیک)
نقص آرژنینو سوکسینات سینتاز
نقص آرژنینو سوکسینیک اسید سینتاز
نقص AS
نقصASS
سیترولینوری
سیترولینمی بزرگسالان گاهی با نام سیترولینمی نوع ۲ و سیترولینمی تاخیری خوانده می شود.
- اسیدوری ناشی از آرژینیوسوکسینات
اسیدوری آرژنینوسوکسینیک یک اختلال ارثی است که باعث تجمع آمونیاک در خون می شود. آمونیاک که هنگام شکسته شدن پروتئین ها در بدن تشکیل می شود، اگر سطح آن خیلی زیاد شود سمی است. سیستم عصبی به ویژه به اثرات آمونیاک اضافی حساس است. اسیدوری آرژنینوسوکسینیک معمولاً در چند روز اول زندگی آشکار می شود. یک نوزاد مبتلا به اسیدوری آرژنینوسوکسینیک ممکن است کمبود انرژی (بی حال) داشته باشد یا تمایلی به خوردن نداشته باشد و سرعت تنفس یا دمای بدنش ضعیف باشد. برخی از نوزادان مبتلا به این اختلال دچار تشنج یا حرکات غیرعادی بدن می شوند یا به کما می روند. عوارض ناشی از اسیدوری آرژنینوسوکسینیک ممکن است شامل تاخیر رشد و ناتوانی ذهنی باشد. آسیب کبدی پیشرونده، فشار خون بالا (فشار خون بالا)، ضایعات پوستی و موهای شکننده نیز ممکن است دیده شود. گاهی اوقات، افراد ممکن است نوع خفیف این اختلال را به ارث ببرند. این افراد می توانند تنها در طول دوره های بیماری یا استرس های دیگر، یا ناتوانی ذهنی خفیف یا ناتوانی های یادگیری بدون شواهدی از افزایش سطح آمونیاک، تجمع آمونیاک در جریان خون داشته باشند.
شیوع: اسیدوری آرژنینوسوکسینیک تقریباً در 1 در 70000 تا 218000 نوزاد رخ می دهد. اکثر موارد این وضعیت بلافاصله پس از تولد توسط غربالگری نوزاد تشخیص داده می شود.
جهش در ژن ASL باعث اسیدوری آرژینینوسوکسینیک می شود. این وضعیت به دسته ای از بیماری های ژنتیکی به نام اختلالات چرخه اوره تعلق دارد، زیرا این بیماری ها ناشی از مشکلات فرآیندی در بدن به نام چرخه اوره هستند. چرخه اوره دنباله ای از واکنش هایی است که در سلول های کبدی رخ می دهد. این چرخه نیتروژن اضافی را که با استفاده از پروتئین توسط بدن ساخته می شود، تجزیه می کند تا ترکیبی به نام اوره بسازد. اوره از طریق ادرار از بدن خارج می شود. شکستن نیتروژن اضافی و دفع آن به صورت اوره از تجمع آن در بدن به صورت آمونیاک جلوگیری می کند. ژن ASL دستورالعمل هایی را برای ساخت آنزیمی به نام آرژنینوسوکسینات لیاز ارائه می دهد که برای مرحله چهارم چرخه اوره مورد نیاز است. نقش ویژه آنزیم آرژنینوسوکسینات لیاز شروع واکنشی است که در آن اسید آمینه آرژنین، یک واحد سازنده پروتئین، از آرژنینوسوکسینات، مولکولی که نیتروژن زائد جمعآوری شده در چرخه اوره را حمل میکند، تولید میشود. آرژنین بعداً به اوره که دفع می شود و اورنیتین که چرخه اوره را دوباره شروع می کند، تجزیه می شود. در افراد مبتلا به اسیدوری آرژنینوسوکسینیک، آرژنینوسوکسینات لیاز ناکارآمد است یا از بین رفته است. در نتیجه، چرخه اوره نمی تواند به طور عادی پیش برود، آرژنین تولید نمی شود و نیتروژن به طور موثر تجزیه نمی شود. نیتروژن اضافی به شکل آمونیاک در خون تجمع می یابد. این تجمع آمونیاک به مغز و سایر بافت ها آسیب می رساند و باعث مشکلات عصبی و سایر علائم و نشانه های اسیدوری آرژینینوسوکسینیک می شود. مشخص نیست که چگونه کمبود آرژنین به ویژگی های این بیماری کمک می کند. این وضعیت در الگوی اتوزومال مغلوب به ارث می رسد، به این معنی که هر دو نسخه از ژن در هر سلول دارای جهش هستند. والدین یک فرد مبتلا به بیماری اتوزومال مغلوب هر کدام یک نسخه از ژن جهش یافته را حمل می کنند، اما معمولاً علائم و نشانه های این بیماری را نشان نمی دهند.
نام های دیگر برای این وضعیت
Argininosuccinate lyase deficiency
Argininosuccinic acidemia
Argininosuccinicaciduria
Argininosuccinyl-CoA lyase deficiency
Arginosuccinase deficiency
ASA
ASAuria
ASL deficiency
درمان های تغذیه ای:
کودک شما ممکن است نیاز به رژیم کم پروتئین داشته باشد تا از خوردن غذاهای خاصی که نمی تواند تجزیه کند، جلوگیری شود. یک متخصص تغذیه می تواند به شما در برنامه ریزی رژیم غذایی مناسب برای رشد سالم کودک، کمک کند.
پزشک همچنین ممکن است فرمول های و غذاهای خاص را برای کودک مبتلا به ASA توصیه کند. این فرمول ها به احتمال زیاد بایستی تا بزرگسالی ادامه یابد.
مکمل ها و داروها:
بسیاری از نوزادان مبتلا به ASA ، مکمل آرژینین دریافت می کنند. آرژینین یک ماده طبیعی است و می تواند به کاهش سطح آمونیاک بالای ناشی از بیماری ASA، کمک کند. تمام افراد مقداری آمونیاک در خون خود دارند، اما سطوح بالای آن، می تواند سمی باشد. پزشک می تواند نسخه ای برای این مکمل ها تجویز کند.
هنگامی که ASA، زود هنگام درمان شود، کودکان می توانند رشد و نمو طبیعی داشته باشند. به همین دلیل غربالگری ASA، بسیار مهم است. غربالگری نوزادان می تواند قبل از اینکه سطح آمونیاک در خون نوزاد به حد خطرناک بالا برسد، فرصت درمان دهد.
بعضی از کودکان حتی پس از درمان، همچنان میزان آمونیاک خونشان بالاست. این کودکان ممکن است نیاز به درمان در یک بیمارستان برای حذف آمونیاک از خون خود داشته باشند.
درمان ASA بسیار مهم است. کودکانی که درمان را دریافت نمی کنند، در معرض معلولیت های ذهنی ، تأخیر رشد، آسیب کبدی، آسیب مغزی، کما یا مرگ می باشند.
وقتی غذا می خوریم، آنزیم ها آن را تجزیه می کنند. برخی از آنزیم ها پروتئین ها را به واحدهای ساختمانی خود تجزیه می کنند که اسید آمینه نامیده می شوند. آنزیم های دیگر این اسیدهای آمینه را به مولکول های بازی خود تجزیه می کنند. هنگامی که پروتئین ها را تجزیه می کنیم، بدن ما نیاز به آنزیم های بیشتری برای رها شدن از مواد زائد دارد. در آرژینینوسوکسینیک اسیدوری، آنزیم آرژینینوسوکسینیک اسید لیاز (ASAL) به درستی کار نمی کند. عمل ASAL این است که به از بین بردن آمونیاک، با دفع آن در ادرار، کمک می کند. آمونیاک یک ماده زائد حاصل از تجزیه پروتئین است.
کودکان مبتلا به آرژینینوسوکسینیک اسیدوری (ASA) یا به اندازه کافی آنزیم آرژینینوسوکسینیک اسید لیاز تولید نمیکنند یا نوع آنزیم غیرفعال را تولید میکنند. هنگامی که ASAL به درستی عمل نکند، بدن نمی تواند از طریق ادرار، آمونیاک را حذف کند. این امر سبب تجمع آمونیاک در بدن می شود.
Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome
در سندرم هیپراورنیتینمی-هیپرآمونمی-هموسیترولینوریا وضعیتی است که در آن بدن قادر به پردازش و حذف مواد زائد، آمونیاک نیست. هنگامی که روند دفع مواد زائد در بدن مختل می شود، مقادیر خطرناک آمونیاک در خون شروع به انباشته شدن می کند. در صورت عدم درمان، این امر می تواند منجر به تاخیر رشد، ناتوانی در یادگیری یا سفتی ناشی از کشش غیر طبیعی عضلات (اسپاستیسیته) شود. تشخیص زودهنگام از طریق غربالگری و درمان نوزاد می تواند از بسیاری از علائم و پیامدهای جدی HHH جلوگیری کند.
سندرم هیپراورنیتینمی-هیپرآمونمی-هموسیترولینور
سندرم گزارش شده است. یک اختلال بسیار نادر است. کمتر از 100 فرد مبتلا در سراسر جهان به این
اسامی دیگر بیماری
- HHH
- Mitochondrial Ornithine Carrier
- Ornithine Translocase Deficiency
- Hyperornithinemia-hyperammonemia-homocitrullinemia syndrome
- Triple H syndrome
سندرم هیپراورنیتینمی-هیپرآمونمی-هموسیترولینوریا (HHH) از نظر شدت و سن شروع آن بسیار متفاوت است. برخی از نوزادان اندکی پس از تولد علائم HHH را نشان می دهند. سایر افراد مبتلا به HHH ممکن است هیچ علامت یا نشانه ای را تا اواخر زندگی نشان ندهند. بسیاری از این علائم ممکن است زمانی رخ دهند که کودک شما غذاها یا شیر خشک هایی را می خورد که بدنش نمی تواند آنها را تجزیه کند. آنها می توانند در مدت طولانی بدون غذا خوردن، بیماری ها و عفونت ها تحریک شوند.
هنگامی که ما غذا می خوریم، بدن ما پروتئین ها را به شکلی تجزیه می کند که می تواند توسط سلول های ما استفاده شود. در طی این فرآیند، یک محصول زائد به نام آمونیاک تولید می شود. آمونیاک از طریق چندین آنزیم مختلف پردازش و از بدن خارج می شود. در سندرم هیپراورنیتینمی-هیپرآمونمی-هموسیترولینوریا (HHH)، آنزیم اورنیتین ترانسلوکاز به درستی کار نمی کند. نوزادان مبتلا به HHH یا اورنیتین ترانسلوکاز غیرفعال است و یا درست کار نمیکند. وقتی اورنیتین ترانسلوکاز به درستی کار نمی کند، بدن نمی تواند آمونیاک را از طریق ادرار دفع کند. این امر باعث تجمع خطرناک آمونیاک در بدن می شود. HHH یک بیماری ژنتیکی اتوزومال مغلوب است. این به این معنی است که یک کودک باید دو نسخه از ژن غیرفعال برای HHH را به ارث ببرد، یکی از هر یک از والدین، تا به این بیماری مبتلا شود. والدین یک کودک مبتلا به بیماری اتوزومال مغلوب هر کدام یک کپی از ژن غیر فعال را دارند، اما معمولاً علائم و نشانههای این بیماری را نشان نمیدهند. در حالی که داشتن یک کودک مبتلا به HHH نادر است، زمانی که والدین هر دو ناقل هستند، می توانند بیش از یک فرزند مبتلا به این بیماری داشته باشند.
علائم HHH عبارتند از:
خستگی
سفتی عضلانی (اسپاستیسیته)
کنترل ضعیف دمای بدن یا تنفس
امتناع از خوردن
استفراغ
تاخیر در رشد
کند شدن رشد
تشنج (همچنین به عنوان صرع شناخته می شود)
مشکلات هماهنگی
نوزادانی که به دلیل سندرم هیپراورنیتینمی-هیپرآمونمی-هموسیترولینوریا (HHH) تحت درمان قرار می گیرند، می توانند رشد و تکامل سالمی داشته باشند. به همین دلیل است که غربالگری نوزادان برای HHH بسیار مهم است. برخی از کودکان حتی با درمان هنوز سطح آمونیاک بالایی در خون خود دارند. این افراد ممکن است نیاز به درمان در بیمارستان برای حذف آمونیاک از خون خود داشته باشند. شناسایی و درمان زودهنگام HHH بسیار مهم است زیرا کودکانی که درمان HHH دریافت نمی کنند در معرض خطر ناتوانی های یادگیری، تشنج، آسیب مغزی یا کما هستند.
- نقص آنزیم اورنیتین ترانس کربامیلاز
کمبود اورنیتین ترانس کاربامیلاز یک اختلال ارثی است که باعث تجمع آمونیاک در خون می شود. آمونیاک که هنگام شکسته شدن پروتئین ها در بدن تشکیل می شود، اگر سطح آن خیلی زیاد شود سمی است. سیستم عصبی به ویژه به اثرات آمونیاک اضافی حساس است. کمبود اورنیتین ترانس کاربامیلاز می تواند در هر سنی آشکار شود. شدیدترین شکل آن در چند روز اول زندگی رخ می دهد. این نوع اختلال با شروع نوزادی معمولاً مردان را تحت تأثیر قرار می دهد. در زنان بسیار نادر است. نوزاد مبتلا به کمبود اورنیتین ترانس کاربامیلاز در دوران نوزادی ممکن است کمبود انرژی (بی حال) داشته باشد یا تمایلی به خوردن نداشته باشد و سرعت تنفس یا دمای بدنش ضعیف باشد. نوزادان مبتلا به این اختلال ممکن است به عنوان "فلاپی" توصیف شوند و ممکن است تشنج یا کما را تجربه کنند. عوارض ناشی از کمبود اورنیتین ترانس کاربامیلاز ممکن است شامل تاخیر رشد و ناتوانی ذهنی باشد. آسیب کبدی پیشرونده نیز ممکن است رخ دهد. در برخی از افراد مبتلا، علائم و نشانه های کمبود اورنیتین ترانس کاربامیلاز ممکن است کمتر شدید باشد و ممکن است تا اواخر زندگی ظاهر نشود. شکل دیررس این اختلال در مردان و زنان رخ می دهد. افراد مبتلا به کمبود دیررس اورنیتین ترانس کاربامیلاز ممکن است دوره هایی از تغییر وضعیت ذهنی مانند هذیان، رفتار نامنظم یا کاهش سطح هوشیاری را تجربه کنند. سردرد، استفراغ، بیزاری از غذاهای پروتئینی و تشنج نیز می تواند در این شکل از اختلال رخ دهد. در این بیماری چرخه اوره مختل شده وآمونیاک به ویژه به سیستم عصبی آسیب می رساند، بنابراین کمبود اورنیتین ترانس کاربامیلاز باعث مشکلات عصبی و همچنین آسیب نهایی به کبد می شود.
شیوع: برآوردها از شیوع کمبود اورنیتین ترانس کاربامیلاز از 1 در 14000 تا 1 در 77000 نفر متغیر است. افراد مبتلا به این اختلال با شروع نوزادی بیشتر در این تخمین ها به حساب می آیند، زیرا افراد مبتلا به شکل دیررس کمتر به مراقبت های پزشکی مراجعه می کنند..
جهش درژنOTC باعث کمبود اورنیتین ترانس کاربامیلاز می شود. ژن OTC دستورالعمل هایی را برای ساخت آنزیم اورنیتین ترانس کاربامیلاز ارائه می دهد.
اسیدوری ارگانیک
در این زیر گروه به انواعی از بیماری همانند موارد زیر می توان اشاره نمود که متعاقبا تمامی آنها در صفحات مجزا شرح داده شده اند:
- گلوتاریک اسیدوری تیپ1
گلوتاریک اسیدمی نوع Iهمچنین به نام اسیدوری گلوتاریک نوع1 یک اختلال ارثی است که در آن بدن قادر به پردازش پروتئین های خاص به درستی نیست. این بیماری به عنوان یک اختلال اسید آلی طبقه بندی می شود، که وضعیتی است که منجر به تجمع غیر طبیعی اسیدهای خاص به نام اسیدهای آلی می شود. سطوح غیر طبیعی اسیدهای آلی در خون (اسیدمی ارگانیک)، ادرار (اسیدوری آلی) و بافت ها می تواند سمی باشد و می تواند باعث مشکلات جدی سلامتی شود. افراد مبتلا به اسیدمی گلوتاریک نوع I دارای سطوح ناکافی آنزیمی هستند که به تجزیه اسیدهای آمینه لیزین، هیدروکسی لیزین و تریپتوفان که بلوک های سازنده پروتئین هستند، کمک می کند. سطوح بیش از حد این اسیدهای آمینه و محصولات تجزیه میانی آنها می تواند تجمع یافته و به مغز آسیب برساند، به ویژه گانگلیون های پایه، که مناطقی هستند که به کنترل حرکت کمک می کنند. ناتوانی ذهنی نیز ممکن است رخ دهد. برخی از نوزادان مبتلا به اسیدمی گلوتاریک نوع I با سرهای غیرعادی بزرگ (ماکروسفالی) متولد می شوند. افراد مبتلا ممکن است در حرکت مشکل داشته باشند و ممکن است دچار اسپاسم، تکان خوردن، سفتی یا کاهش تون عضلانی شوند. برخی از افراد مبتلا به اسیدمی گلوتاریک دچار خونریزی در مغز یا چشم شده اند که ممکن است با اثرات سوء استفاده از کودک اشتباه گرفته شود. کنترل شدید رژیم غذایی ممکن است به محدود کردن پیشرفت آسیب عصبی کمک کند. استرس ناشی از عفونت، تب یا سایر نیازهای بدن ممکن است منجر به بدتر شدن علائم و نشانه ها شود و تنها بهبودی نسبی داشته باشد.
گلوتاریک اسیدمی نوع 1 در هر 100000نفر رخ میدهد. این بیماری در جامعه آمیش ها و در جمعیت اوجیبوا در کانادا بسیار شایع تر است، جایی که از هر 300 نوزاد ممکن است 1 نفر مبتلا شود.
جهش در ژن GCDH باعث اسیدمی گلوتاریک نوع I می شود. ژن GCDH دستورالعمل هایی را برای ساخت آنزیم گلوتاریل-CoA دهیدروژناز ارائه می دهد. این آنزیم در پردازش اسیدهای آمینه لیزین، هیدروکسی لیزین و تریپتوفان نقش دارد. جهش در ژن GCDH از تولید آنزیم جلوگیری می کند یا منجر به تولید آنزیم معیوب می شود که نمی تواند کار کند. کمبود این آنزیم به لیزین، هیدروکسی لیزین و تریپتوفان و محصولات تجزیه میانی آنها اجازه می دهد تا به سطوح غیر طبیعی افزایش یابند، به خصوص در مواقعی که بدن تحت استرس است. محصولات تجزیه میانی ناشی از پردازش ناقص لیزین، هیدروکسی لیزین و تریپتوفان می توانند به مغز، به ویژه گانگلیون های پایه آسیب وارد کنند و باعث علائم و نشانه های اسیدمی گلوتاریک نوع I شوند.
این وضعیت در الگوی اتوزومال مغلوب به ارث می رسد، به این معنی که هر دو نسخه از ژن در هر سلول دارای جهش هستند. والدین یک فرد مبتلا به بیماری اتوزومال مغلوب هر کدام یک نسخه از ژن جهش یافته را حمل می کنند، اما معمولاً علائم و نشانه های این بیماری را نشان نمی دهند.
نام های دیگر این اختلال:
GA I
Glutaric acidemia I
Glutaric acidemia type 1
Glutaric aciduria I
Glutaryl-CoA dehydrogenase deficiency
- متیل مالونیک اسیدوری
پروتئینها همراه با غذا وارد بدن ما میشوند، برای این که بدن بتواند از این پروتئینها استفاده کند، باید آنها را به اجزای کوچکتری که اسید آمینه نامیده میشوند، تبدیل کند. سپس آنزیمهای ویژه ای، تغییراتی را در این اسیدهای آمینه ایجاد میکنند تا بدن بتواند از آنها استفاده کند.همچنین چربیهای موجود در مواد غذایی، توسط آنزیمهای خاصی به اسیدهای چرب تبدیل میشوند. متیل مالونیک اسیدمی وقتی به وجود میآید که این آنزیمهای خاص، یا به درستی عمل نکنند و یا اصلاً وجود نداشته باشند. وظیفه این آنزیمها، تغییر اسیدهای آمینه و اسیدهای چرب در جهتی است که بتوانند مورد استفاده بدن قرار گیرند.
افراد مبتلا به (MMA) Methyl Malonic Academia ، برای هضم برخی اسیدهای آمینه و اسیدهای چرب موجود در غذای روزانه خود، با مشکل مواجه می شوند و هنگامی که این آنزیمها به درستی عمل نکنند، سطح موادی که گلایسین و متیل مالونیک اسید نامیده میشوند به همراه دیگر مواد مضر در خون و ادرار بالا رفته و ایجاد مشکل میکنند.
انواع مختلفMMA:
* برخی فرمها با تزریق ویتامین B12 درمان می شوند که به این فرمها، درمان پذیر با ویتامین B12 می گویند. دو فرم از متیل مالونیک اسیدمی که اغلب با ویتامین B12 درمان می شوند عبارتند از نقص کوبالامین A و نقص کوبالامین B
* برخی از فرمهای متیل مالونیک اسیدمی با ویتامین B12 درمان نمی شوند و به عبارتی درمان ناپذیر با ویتامین B12 هستند. یکی از این فرمها Mut 0 نامیده می شود. این بیماری به علت عدم حضور یک آنزیم خاص به نام متیل مالونیک کوآ موتاز (MCM) ، ایجاد می شود.
* فرم دیگر متیل مالونیک اسیدمی که به درمان با ویتامین B12 پاسخ نمی دهد ، Mutنام دارد. در افراد مبتلا به این بیماری آنزیم MCM فعالیت خیلی کمی دارد.
* متیل مالونیک اسیدمی همراه با سیستینوری، فرم دیگری از انواع متیل مالونیک اسیدمی می باشد.
کنترل ساخت آنزیمها در بدن به عهده ژنها است. افراد مبتلا به MMA یک جفت ژن معیوب دارند که به درستی عمل نمیکنند. به خاطر این تغییر ایجاد شده در ژنها، یا آنزیم ساخته نمیشود و یا آنزیم ساخته شده به درستی فعالیت نمیکند.
این کودکان هر یک از ژنهای معیوب را از یکی از والدین به ارث برده اند. به این نوع توارث، وراثت اتوزومال مغلوب می گویند.
در والدین این کودکان، به ندرت اختلالی مشاهده می شود. اما هر یک از والدین یک تک ژن معیوب برای MMA دارد. این والدین ناقل نامیده می شوند. ناقلین علامت بیماری را بروز نمی دهند زیرا دیگر ژن آنها سالم است.
وقتی هر دو والدین ناقل باشند، ۲۵% احتمال تولد کودک کاملا سالم،۵۰% احتمال تولد کودک ناقل همچون والدین و ۲۵% احتمال تولد کودک مبتلا به MMA در هر بارداری وجود دارد.
مشاوران ژنتیک برای پاسخگویی به سوالات خانواده ها در دسترس می باشند. این مشاوران نحوه توارث بیماری را توضیح خواهند داد و همچنین راهنمای خانواده ها برای بارداری بعدی و همچنین مشاورآنها برای انجام تستهای تشخیصی برای دیگر اعضای خانواده خواهند بود.
این بیماری در دختران و پسران به یک نسبت مشاهده می شود.
امکان انجام تست ژنتیک یا آزمایش DNA جهت بررسی تغییرات ایجاد شده در ژنها وجود دارد. در صورت هرگونه ابهام درباره آزمایش ژنتیک با پزشک متخصص بیماریهای متابولیک و یا با مشاور ژنتیک خود مشورت کنید. ممکن است، انجام آزمایش ژنتیک برای تشخیص بیماری ضروری نباشد، اما اگر امکان آن وجود داشته باشد، میتواند برای تشخیص قبل از تولد و یا تشخیص ناقلین مفید باشد.
اگر یکی از فرزندان شما مبتلا به MMA است، شما میتوانید آزمایش DNA را در طول بارداری بعدی انجام دهید. این آزمایش به شما خواهد گفت که، کودک بعدی شما مبتلا به این بیماری هست یا خیر. نمونه مورد نیاز برای انجام این تست از آمینوسنتز و یا CVS به دست میآید. این بیماری همچنین به وسیله تستهای آنزیمی که بر روی سلولهای بدن جنین انجام میشود، قابل شناسایی میباشد. نمونه مورد نیاز برای این تست از آمینوسنتز به دست میآید. والدین میتوانند تصمیم بگیرند که، تستهای آزمایشگاهی را در طول بارداری انجام دهند و یا بعد از تولد کودک، تستها را روی نوزاد انجام دهند. مشاوران درباره این که کدام روش بهتر است به والدین توضیح خواهند داد.
علایم بیماری:
متیل مالونیک اسیدمی در کودکان اثرات متفاوتی دارد. بسیاری از کودکان مبتلا، علایم بیماری را در اولین روزهای زندگی خود نشان میدهند، در حالی که برخی دیگر ممکن است علایم بیماری را چند سال بعد بروز دهند. در برخی از بیماران هیچ علامتی مشاهده نمیشود. در بیماران متیل مالونیک اسیدمی حملات ناگهانی و پیش بینی نشده بیماری دیده میشود. این حملات ناگهانی، بحرانهای متابولیکی نامیده میشوند.
علایم بحرانهای متابولیکی عبارتند از: کم اشتهایی، استفراغ، احساس ضعف و خواب آلودگی، سستی ماهیچهها و مفاصل.
مشکلات ناشی از عدم کنترل بیماری و بحرانهای متابولیکی عبارتند از: مشکلات تنفسی، تشنج، سکته و کُما که ممکن است منجر به مرگ شوند.
- وجود کتونها در ادرار
- افزایش سطح گلایسین در خون و ادرار
- افزایش سطح برخی مواد سمی در خون
- افزایش مواد اسیدی خون یا اسیدوز متابولیکی
- افزایش متیل مالونیک اسید در خون و ادرار
- کاهش پلاکت خون
- افزایش آمونیاک خون
- افزایش پروپیونیک اسید در خون و ادرار
- کاهش گلبول های سفید خون
- کم خونی
تأثیرات بلند مدت MMA
این عوارض در برخی از کودکان دیده میشود و شامل موارد زیر است:
- ناتوانی در یادگیری و یا عقبماندگی ذهنی
- انعطاف پذیری کم ماهیچهها
- استئوپروزیس (پوکی استخوان)
- اختلال در راه رفتن و دیگر مهارتهای حرکتی
- عدم رشد
- نارسایی کلیه یا از کار افتادگی کلیه
- حرکات غیر ارادی و غیر طبیعی
- بزرگی کبد
- راشهای پوستی
درمان
پزشک کودک شما با یک پزشک متخصص بیماریهای متابولیک و یک متخصص تغذیه همکاری خواهد کرد تا کودک شما را درمان کند. برای جلوگیری از عقبماندگی ذهنی و مشکلات جسمی درمان فوری نیاز است.
در ادامه روش درمانی که به طور معمول برای بیماران MMA پیشنهاد میشود، ذکر شده است:
تجویز دارو
روش اصلی درمان، در کودکانی که به درمان با ویتامین B12 پاسخ میدهند، تزریق ویتامین B12 به صورت هیدروکسی کوبالامین یا سیانو کوبالامین است. تزریق ویتامین B12 میتواند از بروز علایم بیماری در این افراد جلوگیری کند.
برای کودکان مبتلا به MMA استفاده از L-Carnitine میتواند مفید باشد. این دارو یک ماده طبیعی مفید است که به تولید انرژی در بدن کمک میکند. همچنین به بدن کمک میکند تا از مواد مضر رهایی یابد. پزشک به شما خواهد گفت که آیا کودک شما به آن نیازمند است، یا خیر.
برخی آنتی بیوتیک های خوراکی میتوانند به کاهش متیل مالونیک اسید در روده کمک کنند. پزشک به شما خواهد گفت که آیا کودکتان به آنتی بیوتیک خاصی نیاز دارد یا خیر.
رژیم غذایی کم پروتئین، غذاهای طبی و شیر خشکهای خاص
یک برنامه غذایی با مقادیر کم اسیدهای آمینه لوسین، والین، متیونین و ترئونین و با حداقل مقدار پروتئین توصیه میشود. اغلب مواد غذایی موجود در رژیم، کربوهیدراتها (نان، غلات، ماکارونی و میوهها و سبزیجات و غیره) خواهد بود. کربوهیدراتها انواع مختلفی از قندها را به بدن میدهند، که بدن این قندها را صرف تولید انرژی خواهد کرد. رژیم غذایی با کربوهیدرات زیاد و پروتئین کم میتواند از بحران متابولیکی جلوگیری کند.
غذاهایی که حاوی پروتئین بالا بوده و باید از رژیم حذف شده و یا مقدار آن محدود شوند شامل موارد زیر میشود:
شیر و ترکیبات لبنی، گوشت و مرغ، ماهی، تخم مرغ، سبزیجات خشک شده، باقلا خشک شده و مغزها (بادام، پسته، گردو و غیره).
بسیاری از میوهها و سبزیجات مقدار کمی پروتئین دارند و میتوانند به مقدار معینی استفاده شوند. تمام پروتئین را از رژیم غذایی کودک حذف نکنید. کودکان MMA به مقداری پروتئین برای رشد مناسب نیازمندند.
- پروپیونیک اسیدوری
پروتئینها همراه با غذا وارد بدن ما می شوند، برای اینکه بدن بتواند از این پروتئینها استفاده کند، آنها باید به اجزا کوچکتری که اسید آمینه نامیده می شوند، تبدیل شوند. سپس آنزیمهای ویژه، تغییراتی را در این اسیدهای آمینه ایجاد می کنند تا بدن بتواند از آنها استفاده کند.پروپونیک اسیدمی وقتی به وجود می آید که آنزیم ویژه ای که پروپیونیل کوآ کربوکسیلاز (PCC) نامیده میشود، یا به درستی عمل نکند و یا اصلا وجود نداشته باشد. وظیفه این آنزیم، تغییراسیدهای آمینه در جهتی است که بتوانند مورد استفاده بدن قرار گیرند. هنگامی که این آنزیم به درستی عمل نکند سطح برخی مواد، که گلایسین و پروپیونیک اسید نامیده می شوند به همراه دیگر مواد مضر در خون بالا رفته و ایجاد مشکل می کنند.چهار اسیدآمینه ای که به درستی تجزیه نمی شوند عبارتند از: ایزو لوسین ، والین ، متیونین و ترئونین. این اسیدهای آمینه در تمامی غذاهای حاوی پروتئین یافت می شوند. این پروتئینها در گوشت و تخم مرغ و دیگر محصولات لبنی به مقدار بیشتر و در آرد و غلات و برخی میوه ها و سبزیجات به مقدار کمتر یافت می شوند.
کنترل ساخت آنزیمها در بدن به عهده ژنها است. افراد مبتلا به PA یک جفت ژن معیوب دارند که به درستی عمل نمی کنند. به خاطر این تغییر ایجاد شده در ژنها، یا آنزیم PCC ساخته نمی شود و یا آنزیم ساخته شده به درستی فعالیت نمی کند.
پروپونیک اسیدمی در کودکان اثرات متفاوتی دارد. بسیاری از کودکان مبتلا، علایم بیماری را در اولین روزهای زندگی خود نشان می دهند، در حالیکه برخی دیگر ممکن است این علایم را چند سال بعد بروز دهند. در بیماران پروپونیک اسیدمی حملات ناگهانی و پیش بینی نشده بیماری، دیده میشود. این حملات ناگهانی بحرانهای متابولیکی نامیده می شوند.
علایم بحرانهای متابولیکی عبارتند از:
کم اشتهایی
استفراغ
احساس ضعف و خواب آلودگی
سستی ماهیچه ها و مفاصل
عمومی ترین یافته های آزمایشگاهی عبارتند از:
وجود کتونها در ادرار افزایش سطح برخی ارگانیک اسیدها
افزایش سطح گلایسین
افزایش مواد اسیدی خون یا اسیدوز متابولیکی کاهش گلبولهای سفید خون
افزایش آمونیاک خون، کاهش پلاکت خون
مشکلات ناشی از عدم درمان بحرانهای متابولیکی عبارتند از:
مشکلات تنفسی
تشنج
تورم مغز
سکته وکوما که ممکن است منجر به مرگ بیمار شود
بین حملات بحرانهای متابولیکی، کودک اغلب بدون مشکل است.
دلایل احتمالی ایجاد بحران متابولیکی:
دریافت مقدار زیادی پروتئین
بیماری و یا عفونت
گرسنگی طولانی
رویدادهای استرس زا همچون عمل جراحی
تاثیرات بلند مدت PA که در برخی از کودکان دیده می شود شامل موارد زیر است:
ناتوانی در یادگیری و یا عقب ماندگی ذهنی انعطاف پذیری کم ماهیچه ها استئوپروزیس ( پوکی استخوان )
اختلال در راه رفتن و دیگر مهارتهای حرکتی عدم رشد التهاب پانکراس یا پانکراتیتیس
حرکات غیر ارادی و غیر طبیعی، تشنج، راشهای پوستی
اگر درمان دراین بیماران انجام نشود، آسیبهای مغزی به وقوع می پیوندد که می تواند باعث عقب ماندگی ذهنی کودک و یا حتی مرگ او در چند سال اول زندگی شود. تعداد کمی از افراد مبتلا، هیچگاه علایم بیماری را بروز نمی دهند. این افراد هنگامی شناسایی می شوند که یکی از اعضای خانواده آنها که مبتلا به این بیماری است، شناسایی شود.
کودکانی که درمان فوری و مداوم قبل از بحران متابولیکی دارند، ممکن است رشد طبیعی داشته باشند. به طور کلی هر چه درمان زودتر آغاز شود نتیجه بهتری خواهد داد. در برخی از کودکان علیرغم شروع درمان، علایم عقب ماندگی ذهنی و مشکلات یادگیری دیده می شود.همچنین تشنج و مشکلات رفتاری در برخی از کودکان گزارش شده است. کودکان مبتلا به پروپونیک اسیدمی اغلب بیشتر از معمول دچار عفونت می شوند، که این عفونتها باید هر چه سریعتر درمان شوند تا به بحران متابولیکی منجر نشوند.
ژیم غذایی کم پروتئین، غذاهای طبی و شیر خشکهای خاص:
یک برنامه غذایی با مقادیرکم اسیدهای آمینه لوسین، والین، متیونین و ترئونین و با حداقل مقدار پروتئین توصیه می شود. اغلب مواد غذایی موجود دراین رژیم، کربو هیدراتها (نان، غلات، میوه ها و سبزیجات و غیره) خواهد بود.کربو هیدراتها انواع مختلفی از قندها را به بدن می دهند، که بدن این قندها را صرف تولید انرژی خواهد کرد. رژیم غذایی حاوی کربو هیدرات زیاد و پروتئین کم، می تواند از بحران متابولیکی جلوگیری کند.
غذاهایی که حاوی پروتئین بالا بوده و باید از رژیم حذف شده و یا مقدار آنها محدود شوند عبارتند از:
شیر و ترکیبات لبنی، گوشت و مرغ، ماهی، تخم مرغ، سبزیجات خشک شده، باقلا خشک شده و مغزها (بادام، پسته، گردو و غیره)، بسیاری از میوه ها و سبزیجات مقدار کمی پروتئین دارند و می توانند به مقدار معینی استفاده شوند. تمام پروتئین را از رژیم غذایی کودک حذف نکنید. کودکان PA به مقداری پروتئین برای رشد مناسب نیازمندند.
متخصص تغذیه، رژیم غذایی مناسب رشد کودک شما را طراحی خواهد نمود. کودک شما به این رژیم مناسب غذایی، در طول زندگی خود نیازمند خواهد بود. علاوه بر این رژیم غذایی کم پروتئین، ممکن است به کود ک شما شیر خشک های خاص نیز تجویز شود. این شیر خشک ها، حاوی مقادیر مناسب پروتئین و دیگر مواد غذایی لازم برای رشد کودک خواهد بود. پزشکان به شما خواهند گفت که کدام شیر خشک و چه مقدار از آن برای کودک شما مناسب است.
همچنین غذاهای طبی همچون آرد های با پروتئین کم، ماکارونی و برنج ساخته شده اند که مناسب افراد PA می باشند. متخصص تغذیه به شما خواهد گفت که چگونه می توانید از این غذاها در برنامه غذایی کودکتان استفاده کنید.
از گرسنگی طولانی مدت کودک پرهیز کنید:
کودکان مبتلا به PA نیازمندند که به طور متناوب غذا بخورند تا از بحران متابولیکی در آنها جلوگیری شود. اغلب این کودکان باید هر 6-4 ساعت یک بار غذا بخورند. حتی برخی از کودکان ممکن است به وعده های غذایی بیشتری نیاز داشته باشند. کودک شما حتی در طول شب نیز نباید گرسنه بماند. اگر کودکتان در طول شب خودش از خواب بیدار نشود، شما مجبور خواهید شد خودتان او را از خواب بیدار کنید.
ممکن است به شما گفته شود که یک غذای نشاسته ای سبک، یک نوبت قبل از خواب و یک نوبت در طول شب به کودکتان بدهید. ممکن است کودک شما به یک نوبت دیگر از این غذا، هنگام صبح بعد از بیدار شدن از خواب نیاز داشته باشد. نشاسته ذرت خام مخلوط شده با آب، مقدار معین شیر و یا دیگر نوشیدنیها ممکن است یک منبع انرژی مناسب برای کودکانPA باشد که گاهی برای کودکان بالاتر از یک سال پیشنهاد می شود. در مورد غذای نشاسته ای مناسب با متخصص تغذیه مشورت کنید.
تجویز دارو
برای کودکان مبتلا بهPA استفاده از L-Carnitine می تواند مفید باشد. این دارو یک ماده طبیعی مفید است که به تولید انرژی در بدن کمک می کند همچنین به بدن کمک می کند از مواد مضر رهایی یابد. این دارو بخشی از درمان متداولPA است. پزشک به شما خواهد گفت که کودک شما به چه مقدار از آن نیازمند است. برخی آنتی بیوتیکهای خوراکی می توانند به کاهش مقدار اسید پروپیونیک در روده کمک کنند.
پزشک به شما خواهد گفت که آیا کودکتان به آنتی بیوتیک خاصی نیاز دارد یا خیر. به برخی از کودکان ممکن است مکمل خوراکی بیوتین داده شود. بیوتین یک نوع ویتامین B است که به بدن کمک می کند از غذا انرژی بسازد. تاثیر بیوتین در PA هنوز به اثبات نرسیده است اما ممکن است از شما خواسته شود که آن را به بیمار بدهید تا پزشک بتواند تاثیر این مکمل را در روند درمان کودک مشاهده کند.
کودکانی که علایم بحران متابولیکی را دارند، باید در بیمارستان تحت درمان قرار گیرند. در هنگام بحران متابولیکی کودک شما ممکن است به داروهایی همچون بی کربنات (به صورت داخل وریدی) نیازمند باشد. این دارو به کاهش سطح مواد اسیدی خون کمک می کند. گلوکز نیز اغلب به صورت وریدی به بیمار داده می شود تا از تجزیه پروتئینها و چربیهای ذخیره ای بدن جلوگیری شود. به یاد داشته باشید که هیچ دارویی را بدون مشورت با پزشک استفاده نکنید.
این بیماری در دختران و پسران به یک نسبت مشاهده می شود. هر فرد دارای دو ژن است که دستور ساخت آنزیم PCC را میدهد. در کودکان PA هیچ یک از این دو ژن به درستی عمل نمی کنند. این کودکان هر یک از ژنهای معیوب را از یکی از والدین خود به ارث برده اند. به این نوع توارث، وراثت اتوزومال مغلوب می گویند. در والدین این کودکان، به ندرت اختلالی مشاهده می شود. اما هر یک از والدین یک تک ژن معیوب برای PA دارد. این والدین ناقل نامیده می شوند.
ناقلین علامت بیماری را بروز نمی دهند زیرا دیگر ژن آنها سالم است. وقتی هر دو والدین ناقل باشند، 25% احتمال تولد کودک کاملا سالم،50% احتمال تولد کودک ناقل همچون والدین و 25% احتمال تولد کودک مبتلا به PA در هر بارداری وجود دارد. مشاوران ژنتیک برای پاسخگویی به سوالات خانواده ها در دسترس می باشند. این مشاوران نحوه توارث بیماری را توضیح خواهند داد و همچنین راهنمای خانواده ها برای بارداری بعدی و همچنین مشاور آنها برای انجام تستهای تشخیصی برای دیگر اعضای خانواده خواهند بود.
اگر یکی از فرزندان شما مبتلا بهPA است، شما میتوانید آزمایش DNA را در طول بارداری بعدی انجام دهید. این آزمایش به شما خواهد گفت که، کودک بعدی شما مبتلا به این بیماری هست یا خیر. نمونه مورد نیاز برای انجام این تست از آمینوسنتز و یا CVS به دست می آید. این بیماری همچنین به وسیله تستهای آنزیمی که بر روی سلولهای بدن جنین انجام می شود، قابل شناسایی می باشد.
نمونه مورد نیاز برای این تست از آمینوسنتز به دست می آید. والدین میتوانند تصمیم بگیرند که، تستهای آزمایشگاهی را در طول بارداری انجام دهند و یا بعد از تولد کودک، تستها را روی نوزاد انجام دهند. مشاوران درباره این که کدام روش بهتر است به والدین توضیح خواهند داد.
شیوع: شیوع این بیماری در ایالت متحده آمریکا در حدود، 1 مورد درهر 100000 تولد است. پروپونیک اسیدمی در تمامی نژادهای قومی سرتاسر جهان وجود دارد، اما بیشتر از همه در اعراب عربستان سعودی مشاهده می شود. درصد افراد مبتلا در این کشور از یک در 2000 تا 1 در 5000 متغییر است.
نام های دیگر این اختلال
Propionyl-CoA carboxylase deficiency
PCC defect
Glycinemia ketonia
Hyperglycinemia ketonia
- متیل گلوتاکونیک اسیدوری (سندرم بارت)
سندرم بارت یک بیماری ژنتیکی است که عمدتا جنس مذکر را درگیر می کند. برخی از علائم این بیماری شامل بزرگ شدن قلب، کم شدن تعداد سلول های خون، ضعف عضلات و خستگی است. علاوه بر این، می تواند سطح مواد شیمیایی مانند 3- متیگلوتاکونیک اسید و 2-اتیل هیدراکرلیک اسید در ادرار یا خون افزایش یابد. سندرم بارت در اثر تغییرات (جهش) در ژن TAZ ایجاد می شود و دارای الگوی وراثتی وابسته به X است.
سندرم بارت عمدتا در اوایل نوزادی یا کودکی مشاهده می شود. با این حال، در برخی از بیماران، علائم در بزرگسالی ظاهر می شوند. علائم می تواند متفاوت باشد و از یک فرد به فرد دیگر متفاوت است. مردان مبتلا به سندرم بارت می توانند مشکلات قلبی مختلفی مانند موارد زیر را داشته باشند:
- کاردیومیوپاتی متسع ( بزرگ شدن و ضعیف شدن قلب )
- کاردیومیوپاتی هیپرتروفیک (دیواره بین بطنی به طرف چپ ضخیم تر شده و راه خروجی آئورت را مسدود می کند.)
- فیبروالاستوز اندوکارد ( اختلال بافت کلاژن)
- عدم تراکم بطن چپ
کاردیومیوپاتی متسع هنگامی است که عضله بطن چپ بزرگ و ضعیف می شود و این باعث کاهش توانایی قلب در پمپاژ خون می شود. در برخی از افراد مبتلا به سندرم بارت، عضلات قلب بسیار ضخیم می شوند و پمپاژ خون را دشوار می کنند (کاردیومیوپاتی هیپرتروفیک).
گاهی اوقات، این ضخیم شدن ممکن است به دلیل تجمع بافت های همبند و الیاف الاستین (فیبرولاستوز غدد درون ریز) باشد.
در سایر بیماران، بطن های چپ به درستی رشد نمی کنند (عدم تراکم بطن چپ)، بنابراین به جای صاف بودن، عضله ضخیم و اسفنجی می شود و باعث می شود خون رسانی دشوار شود. این یافته های قلبی تقریبا همیشه قبل از 5 سالگی وجود دارد.
گاهی اوقات مشکلات قلبی را می توان در معاینه سونوگرافی در سه ماهه آخر بارداری مشاهده کرد. علاوه بر تفاوت های ساختاری در قلب، در برخی از نوجوانان و جوانان می تواند ضربان قلب نامنظمی (آریتمی) شناسایی شود.
مشکلات قلبی ممکن است منجر به کاهش گردش خون بدن و ریه ها (نارسایی قلبی) شود. علائم نارسایی قلبی ممکن است شامل تنگی نفس، خستگی و حالت تهوع باشد، اما علائم به کودک و سایر عوامل بستگی دارد.
افراد مبتلا به سندرم بارت سطح پایین گلبول های سفید خون (نوتروپنی) دارند. گلبول های سفید بدن در مقابله با عفونت ها به ما کمک می کنند. به دلیل نوتروپنی، افراد دچار زخم دهان، ذات الریه یا عفونت خون هستند.
مردان مبتلا به سندرم بارت دارای عضلات ضعیف (هیپوتونی) به ویژه در دست و پا هستند. به دلیل هیپوتونی، مهارت های حرکتی ناخوشایند مانند خزیدن، نشستن یا راه رفتن در کودکان بیشتر طول می کشد.
به دلیل مشکلات قلبی و عضلات ضعیف، پسران مبتلا ورزش را به خوبی تحمل نمی کنند. مردان مبتلا به این بیماری در دوران کودکی تاخیر رشد دارند، اما در دوره بلوغ رشد قابل توجهی وجود دارد.
علائم دیگر شامل انحنای ستون فقرات (اسکولیوز) و تاخیر در سن استخوان است.
مردان مبتلا به سندرم بارت دارای ویژگی های مشخصی در صورت هستند. آنها صورت گرد با چانه برجسته و گونه های پر دارند. گوش ها بزرگ هستند و چشم هایشان فرورفته است.
ویژگی های صورت با افزایش سن کمتر مورد توجه قرار می گیرند. ویژگی بارز در نوجوانی و بزرگسالی توزیع چربی در باسن، ران و قفسه سینه است.افرادی که مبتلا به این بیماری هستند، نوعی از ناتوانی یادگیری را دارند. آنها مهارت خواندن و واژگان متناسب با سن را دارند. با این حال، ممکن است در ریاضیات به کمک بیشتری احتیاج داشته باشند. اولین کلمات یا جملات که به زبان می آورند، می تواند در مقایسه با افراد دیگر به تاخیر بیفتد. این کودکان در ایجاد مهارت هایی مانند خواندن نقشه، تشخیص اشکال و یافتن اشیا در تصویر تاخیر دارند. پسران مبتلا به این بیماری مشکلات تغذیه ای دارند.
داده های ثبت سندرم بارت نشان می دهد که یک سوم پسران مبتلا به این بیماری برای تغذیه نیاز به لوله ای از طریق بینی یا مستقیم به معده دارند. پسران مبتلا به این شرایط ، بسیار بدغذا هستند. غذاهای شور، پنیری و تند از جمله غذاهایی هستند که آنها ترجیح می دهند.
علاوه بر کاردیومیوپاتی، نوتروپنی و تاخیر رشد، در این افراد همچنین سطح مارکرهای بیوشیمیایی نیز افزایش یافته است. افزایش سطح اسید 3-متیگلوتاکونیک و 2-اتیل هیدراکریلیک اسید در ادرار یا خون، علامت شایعی است که برای رسیدن به تشخیص استفاده می شود. با این حال، هیچ علائمی در ارتباط با افزایش سطح این مواد شیمیایی مشاهده نشده است.
سندرم بارت در اثر جهش در ژن TAZ ایجاد می شود. ژن TAZ پروتئینی به نام تافازین تولید می کند.
تافازین به تجزیه چربی به نام کاردیولیپین کمک می کند. کاردیولیپین در غشای داخلی ساختارهایی به نام میتوکندری وجود دارد. میتوکندری ساختاری در سلول است که به تولید انرژی کمک می کند. از دست دادن پروتئین تافازین عمدتا بر اندام هایی که نیاز به انرژی دارند، مانند عضلات قلب و اسکلتی تاثیر می گذارد. با این حال، برای درک اینکه چگونه از دست دادن تافازین منجر به کاردیومیوپاتی و نوتروپنی می شود، تحقیقات بیشتری لازم است.
سندرم بارت به صورت وابسته به X به ارث می رسد. اختلالات ژنتیکی مرتبط با X شرایطی است که توسط ژن غیرطبیعی در کروموزوم X ایجاد می شود و بیشتر در مردان ظاهر می شود. زنانی که دارای ژن غیرطبیعی در یکی از کروموزوم های X خود هستند ناقل آن اختلال هستند. زنان ناقل معمولا علائمی از خود نشان نمی دهند، زیرا زنان دو کروموزوم X دارند و فقط حامل یک ژن غیرطبیعی است. هیچ زنی که ناقل سندرم بارت باشد و علامت دار باشد گزارش نشده است.مردان دارای یک کروموزوم X هستند که از مادرشان به ارث می رسد و اگر یک مرد کروموزوم X را که حاوی یک ژن غیرطبیعی است، به ارث ببرد، به این بیماری مبتلا می شود. ناقلین زن که یک اختلال وابسته به X داشته باشند، در هر بارداری احتمالات زیر وجود خواهد داشت:
• 25٪ احتمال داشتن یک دختر ناقل مانند خودشان
• 25٪ احتمال داشتن یک دختر غیرناقل
• 25٪ احتمال داشتن پسری مبتلا به این بیماری
• 25٪ احتمال داشتن پسری سالم است.
اگر یک مرد مبتلا به اختلال وابسته به X بتواند تولید مثل کند، ژن غیرفعال را به تمام دخترانش که ناقل هستند منتقل خواهد کرد. یک مرد نمی تواند یک ژن مرتبط با X به پسران خود منتقل کند، زیرا مردان همیشه کروموزوم Y خود را به جای کروموزوم X خود به فرزندان پسر می رسانند.
سندرم بارت معمولا در دوران نوزادی یا اوایل کودکی تشخیص داده می شود، اما در برخی از بیماران در سنین بالاتر تشخیص داده شده است. تشخیص بر اساس ارزیابی بالینی، شناسایی یافته های بدنی مشخص، سابقه کامل بیمار و خانواده و انواع آزمایشات تخصصی است.
اگر کسی علائم زیر را داشته باشد، سندرم بارت را در نظر بگیرید:
• مشکلات قلبی مانند کاردیومیوپاتی متسع، کاردیومیوپاتی هیپرتروفیک و عدم تراکم بطن چپ
• افزایش میزان اسید 3-متیل گلوتاکونیک در خون و یا ادرار
• نوتروپنی
• هیپوتونی
• تاخیر رشد
• ویژگی های مشخصه صورت
در برخی از خانواده های مبتلا به سندرم بارت، میزان خطر از دست دادن بارداری متعدد با جنین پسر مشاهده شده است.
آزمایش ژنتیکی مولکولی برای جهش در ژن TAZ تشخیص سندرم بارت را تایید می کند. آزمایش ژن TAZ می تواند به صورت جداگانه یا به عنوان بخشی از یک آزمایش چند ژنی انجام شود.
درمان سندرم بارت به علائم خاصی که وجود دارد، بستگی دارد. چنین درمان هایی ممکن است به تلاش تیمی از متخصصان پزشکی مانند متخصصان اطفال، متخصصان قلب کودکان، متخصصان خون، متخصصان عفونی، فیزیوتراپیست ها، کاردرمانگرها و یا سایر متخصصان نیاز داشته باشد.
نارسایی قلبی و یا عفونت های باکتریایی تهدیدهای یک بیمار مبتلا به سندرم بارت است. این مشکل یکی از دلایل اصلی کاهش امید به زندگی است. از داروهای استاندارد نارسایی قلبی مانند مسدود کننده های بتا، مهار کننده های ACE و دیگوکسین استفاده می شود. این درمان به بهبود عملکرد قلب کمک می کند و علائم نارسایی قلبی را کاهش می دهد.
از آسپرین برای کاهش تشکیل لخته استفاده می شود. پیوند قلب در صورت نارسایی شدید قلب در نظر گرفته می شود. عملکرد قلب پس از نوزادی بهبود می یابد، بنابراین پیوند قلب باید به دقت مورد توجه قرار گیرد.
برای افراد مبتلا به نوتروپنی تایید شده، با نظارت و شروع درمان زودهنگام عفونت های مشکوک با آنتی بیوتیک، می توان از عوارض ناشی از عفونت باکتریایی جلوگیری کرد. به عنوان مثال، ممکن است آنتی بیوتیک ها به عنوان یک درمان پیشگیرانه در طی نوتروپنی برای جلوگیری از شروع عفونت ارائه شود. مصرف نشاسته ذرت پخته نشده قبل از خواب برای جلوگیری از تحلیل رفتن عضلات توصیه می شود. مداخله زودهنگام مانند فیزیوتراپی برای افزایش قدرت عضلانی توصیه می شود و به کودکان کمک می کند تا به مراحل مختلف رشد برسند. مشاوره ژنتیک برای افراد مبتلا به سندرم بارت و خانواده های آنها توصیه می شود.
متابولیسم اسید های آمینه دارای زنجیره جانبی
در این گروه سه اسید آمینه ضروری بدن یعنی لوسین، ایزولوسین و والین قرار دارند که نقص در متابولیسم هر یک از آنها می تواند منجر به بیماری شربت افرا (Maple Syrup Urine Disease) یا MSUD شود. این بیماری در صفحه اختصاصی خود به تفصیل مورد بحث و بررسی قرار گرفته است که برای اطلاع از آن می توانید اینجا کلیک کنید.
در این بیماری بدن فرد مبتلا نمی تواند بعضی از " اسید های آمینه" را به مصرف برساند (اسید های آمینه از تجزیه پروتئین ها در بدن بدست می آیند). این اسیدهای آمینه عبارتند از: لوسین، ایزولوسین و والین که در طبقه بندی، جزو اسیدهای آمینه شاخه دار (BCAAs یا Branched – chain Amino Acids) دسته بندی می شوند. این سه اسیدآمینه در غذاهای پروتئینی مانند شیر، گوشت و تخم مرغ به وفور و در آرد معمولی، غلات، مغزها (مانند بادام زمینی) و بعضی سبزیجات و میوه ها به مقدارکمتر یافت می شوند.
علت اصلی این بیماری فقدان یا کاهش آنزیمی موسوم به:branched-chain alpha-ketoacid dehydrogen (BCKDH) است. وجود این آنزیم جهت مصرف اسیدهای آمینه فوق وتامین انرژی و رشد کودک ضرورت دارد، وگرنه این مواد در بدن انباشته شده و باعث آسیب به قسمت های مختلف بخصوص مغز ( و درنتیجه تحلیل سیستم عصبی، آنسفالوپاتی، عقب ماندگی ذهنی ) می شوند. ژن این آنزیم در کروموزم 19 قرار دارد.
این انزیم از چهار زیر واحد تشکیل شدهE1α, E1β, E2, and E3)) وقوع موتاسیون در این زیر واحدها موجب بیماری MSUD میشود.
علامت مشخص این بیماری آن است که ادرار مبتلایان بویی شبیه به شربت افرا (بوی نبات یا قند سوخته!) می دهد از این رو "بیماری ادرار با بوی شربت افرا" نام گرفته است.
در حالت طبیعی، کاتابولیسم (=تجزیه) "اسید های آمینه شاخه دار" در کبد، کلیه، عضله، قلب و بافت های چربی انجام می گیرد (که با ورود این اسید آمینه به داخل سلول از طریق ترکیب ناقل غشا سلول آغاز می شود).
در فرد طبیعی واکنش های ابتدایی در مسیر کاتابولیسم لوسین، ایزولوسین و والین مشابه یکدیگرند، ولی بعد از آن، اسکلت کربنی هر یک از این اسیدهای آمینه وارد مسیر منحصر به فردی شده و به ترکیبات واسطه ای متفاوتی تبدیل می شوند (شکل ). کاتابولیسم طبیعی این اسید های آمینه در رشد و تامین انرژی بدن نقش دارند.
بیماری MSUD بر حسب طبقه بندی به پنج گروه تقسیم می شود
1- کلاسیک (classic)
2- حدواسط (Intermediate)
3- متناوب (Intermittent)
4- پاسخ دهنده به تیامین (Thiamine – Responsive)
5- کمبود لیپوآمید دهیدروژناز (LipoAmid Dehydrogenase Deficiency)
1- نوع کلاسیک (Classic): این نوع به دلیل فقدان کمپلکس آنزیمی "آلفا- کتو اسید دهیدروژناز میتوکندریال" رخ داده و شیوع آن 1 در 200000 تولد است. عمومی ترین نوع بیماری است و در آن فعالیت آنزیم صفر یا کم است (فعالیت آنزیم معمولاً کمتر از 2% حد نرمال است).کودکان مبتلا به این شکل از بیماری، در همان چند روز اول عمر علائمی نظیر خوب شیر نخوردن و استفراغ را بروز می دهند و معمولاً قادر به تحمل BCAAs نمی باشند. بنابراین رژیم غذایی آنها باید فاقد این اسیدهای آمینه باشد.
2- واسطه ای (Intermediatبرخلاف فرم کلاسیک سطح فعالیت آنزیمی کمی بالاتر است (حدود 8-3% حد نرمال). این گروه معمولاً مقادیر بیشتری از لوسین را تحمل می کنند. البته در شرایط بیماری و گرسنگی وضعیت کودک شبیه به حالت کلاسیک شده و نوع درمان یکسان خواهد بود.
متناوب (Intermittent): شکل حفیف بیماری است که در آن میزان میزان فعالیت آنزیمی تقریباً 15 – 8% حد نرمال است.3-
در این گروه، کودکان اغلب تا سن 12 تا 24 ماهگی علامتی ندارند ولی در هنگام ناخوشی، گرسنگی، و یا بعد از استرس هایی نظیر جراحی سطح BCAAs در آنها بالا رفته کودک دچار بحران متابولیک (metabolic crisis) می گردد. علائم بحران متابولیک عبارتند از: خواب زیاد، کندی و سستی، استفراغ، بد خلقی و در صورت عدم درمان می تواند باعث سفتی عضلات، تشنج، کوما و حتی مرگ گردد.
4-پاسخ دهنده به تیامین (Thiamine – Responsive) در این نوع مصرف مکمل های حاوی تیامین منجر به افزایش فعالیت آنزیم گشته و در نتیجه به مصرف مصرف لوسین، ایزولوسین و والین کمک می کند. در اغلب موارد تنها محدودیت متوسط پروتئین نیاز است. این نوع در واقع شکل نادرتر بیماری MSUD است.
5- کمبود لیپوآمید دهیدروژناز (LipoAmid Dehydrogenase Deficiency,E3): فرم بسیارنادر بیماری است که با آسیب عصبی، ضعف عضلانی، اختلالات رشد، و مشکلات حرکتی همراه است و در بعضی موارد منجر به مرگ کودک می شود. معمولا کودک تا 6-8 ماهگی علامتی ندارد ولی بعد ازآن در پی ابتلا به وضعیتی موسوم به "اسیدوز لاکتیک " (در اثر تجمع شدید اسید لاکتیک در بدن) علائمی مانند استفراغ، دل درد و تنفس سریع ظاهر می شوند. در صورت عدم درمان می تواند باعث مرگ طفل گردد. در این فرم، علاوه بر اسید لاکتیک، مواد دیگری از جمله اسید های آمینه شاخه دار در بدن تجمع پیدا می کنند.
ژنتیک و نحوه وراثت: MSUD
فردی که دارای یک ژن معیوب (جهش یافته) است "حامل" (carrier)آن ژن نامیده می شود. برای اینکه فردی به بیماری MSUD مبتلا شود، باید پدر و مادر او هر دو حامل ژن معیوب مربوطه باشند، زیرا نحوه وراثت این بیماری از نوع "اتوزوم مغلوب" است.
در حالت طبیعی، در هر فرد دو ژن برای ساخت آنزیم "آلفا کتواسید دهیدروژناز زنجیره جانبی" دخالت دارند که یکی از پدر و دیگری از مادر به وی رسیده است. اگر یکی از آن ژن ها عملکرد نرمال و دیگری عملکرد غیرنرمال داشته باشد، شخص "حامل" ژن بیماری MSUD محسوب می شود و در صورتیکه شخص هر دو ژن معیوب را داشته باشد، "مبتلا" به MSUD محسوب می گردد.
زمانی که پدر و مادر هر دو "حامل" ژن MSUD در هر حاملگی 25% احتمال دارد که جنین هر دو ژن معیوب را دریافت کرده و به بیماری MSUD "مبتلا" شود.احتمال آنکه جنین تنها "حامل" یک ژن برای MSUD باشد (یعنی مثل والدین خود) 50% است.احتمال آنکه جنین ژن های سالم والدین خود را دریافت کند و نرمال باشد، 25% است.
لازم به ذکر است که شانس ابتلا به این بیماری در پسران و دختران به یک اندازه است.
شیوع:
شیوع جهانی این بیماری 1 به 185000 است. در کشور پرتغال 1 به 86800 و در جوامع با ضریب درون همسری بالا (ضریب درون همسری عبارت است از احتمال به ارث بردن یک جفت آلل توسط یک فرد از جد خود از طریق والدین) مثل منونیت ها (Mennonites) در ایالت پنسیلوانیا آمریکا 1 به 200 گزارش شده است.
در مطالعه 10 ساله دکتر زاهد پاشا و همکاران که در سال 92 منتشر شد از مجموع 3154 نوزاد بستری شده در بیمارستان کودکان امیرکلای بابل 16 مورد MSUD تشخیص داده شد که با توجه به کل موالید این استان در این بازه زمانی، میزان شیوع 1 به 26700 گزارش و ناشی از بالا بودن نرخ ازدواج فامیلی (6/38%) و نیز بالا بودن متوسط ضریب درون همسری 0185/0 تلقی گردید (در این مطالعه 5/87% والدین این کودکان رابطه خویشاوندی داشته اند). در این گزارش شیوع 7 برابری بیماری نسبت به آمارهای جهانی قابل توجه است.
علایم:
کودکان مبتلا به فرم کلاسیک بیماری، در هنگام تولد و طی 3 تا 7 روزگی طبیعی به نظر می رسند. کودکان مبتلا به فرم واسطه ای علائمی خفیف تری داشته و بیماری سریع بروز نمی کند.
بطور کلی علائم عبارتند از: نق نق کردن، بی حالی، خوب شیر نخوردن، عدم وزن گیری، هایپوتونیا (شلی عضلات)، هایپرتونیا (سفتی عضلات)، استفراغ، گریه جیغ مانند، حمله ناگهانی بیماری و استشمام بوی مشخص "شربت افرا" از ادرار که هنگام تعویض کهنه بچه پس از خشک شدن، بوی آن شدیدتر هم می شود. اسیدوز، کتوز و از دست دادن آب بدن نیز از علایم دیگر این بیماری است.
همانطوریکه قبلا گفته شد، بارزترین مشخصه این بیماری بوی مخصوص ادرار می باشد، که مشابه بوی شربت درخت افرا یا نبات سوخته است. در این بیماری مقدار لوسین، ایزولوسین و والین و آلفا- کتو اسیدهای مربوط به این ترکیبات در پلاسما و ادرار مبتلایان افزایش می یابد. البته مقادیر کمی آلفا- هیدروکسی اسیدهای شاخه دار که در اثر احیای آلفا- کتو اسید ها تشکیل می شوند نیز در ادرار مبتلایان وجود دارد.
طی غربالگری بیماریهای متابولیک در نوزادان 2 تا 7 روزه با استفاده از تست MS/MS مارکرهای مربوط به اسیدهای آمینه اندازه گیری می شود.
از جمله مارکرهایی که طی این غربالگری اندازه گیری می شوند می توان از اسیدهای آمینه لوسین، ایزولوسین و والین نام برد، که بعنوان مارکرهای اولیه غربالگری MSUD محسوب می گردند. در صورتی که درغربالگری نتیجه تست های فوق بالاتر از حد نرمال باشد، باید تست 1 MS/MS الی 2 روز بعد با خونگیری مجدد، تکرار شود. بدیهی است در صورت وجود مشکل آنزیمی میزان این مارکرها در نمونه دوم افزایش بیشتری پیدا خواهند کرد.
مارکرهای ثانویه: بعد از تائید افزایش مارکرهای اولیه طی دو مرحله نمونه گیری فوق، تغییرات مارکرهای ثانویه طی دو مرحله نمونه گیری نیز با محاسبه نسبت های زیر چک می شود:
افزایش نسبت های لوسین/فنیل آلانین (بیشتر از 7/4)، لوسین/آلانین (بیشتر از 5/1)، ایزولوسین/فنیل آلانین، ایزولوسین/آلانین و والین/فنیل آلانین (بیشتر از 2/5) طی دو مرحله در صورت مشاهده افزایش نسبت های فوق و وجود این تغییرات در مارکرهای ثانویه، جواب فرد به عنوان یک جواب "خارج از محدوده نرمال" تلقی و بعنوان یک تست "مشکوک" از نظر اختلالات اسیدهای آمینه (به ویژه MSUD) گزارش و انجام تست های تائیدی یا تشخیصی برای تائید تشخیص بیماری توصیه می گردد.
تشخیص:
انجام تست پروفایل اسیدهای آمینه ی پلاسما: به صورت افزایش لوسین، ایزولوسین، والین، آلوایزولوسین نمودار می شود. آلانین هم در این بیماران کاهش می یابد. افزایش L- آلوایزولوسین در بیماری MSUD بعنوان مارکر پاتوگنومونیک (قطعاً تشخیصی) محسوب می شود.
انجام تست پروفایل اسیدهای آمینه ی ادرار: به صورت افزایش دفع ادراری اسیدهای آمینه شاخه دار مشخص می شود.
انجام تست پروفایل اسیدهای ارگانیک در ادرار: که به صورت افزایش آلفا کتو اسیدها و هیدروکسی اسیدها نظیر 2-Ketoisocaproate، 2- Ketoisovalerate، 2-Keto-3-methlyvalerate، 2-hydroxyisocaprate، 2-hydroxyisovalerate، 2-hydroxy-3-methylvalerate، phenyllactate و phenylpyrovate خود را نشان می دهد.
مثبت شدن کتون بادیهای ادرار
تست غربالگری 2،4- دی نیتروفنیل هیدرازین در محیط اسیدی: که حضور آلفا کتو اسیدها را در ادرار مشخص می کند. اگر تست مثبت باشد هیدازین به فرم نامحلول در می آید و تست از کدورت خفیف (+1) تا رسوب زرد متمایل به نارنجی (+4) ظاهر می شود. البته این تست در مواردی مثل فنیل کتونوریا (فینل پیروویک اسید)، هیستیدینمیا (ایمیدازول پیرویک اسید)، و سوء جذب متیونین (سندروم Oasthouse ) نیز مثبت می شود.
استشمام بویی شبیه بوی شربت افرا یا نبات سوخته از ادرار
انجام تست های ژنتیکی: امروزه تست های ژنتیکی خاصی وجود دارند (DNA tests) که با کمک آنها می توان در بافت جنین و یا فرد مورد نظر مشخص نمود که آیا حامل ژن MSUD هست یا خیر. (از جمله متدهای تشخیص و غربالگری قبل از تولد میتوان به بررسی نقص فعالیت آنزیم "آلفا کتواسید دهیدروژناز زنجیره جانبی " به کمک کشت پرزهای لایه کوریونیک یا مایع آمینوتیک اشاره کرد).
بررسی ژنتیکی موتاسیون در ژن های BCKDHA، BCKDHB و DBT.
درمان
با شروع درمان در هفته اول تولد می توان تا حد زیادی جلوی عواقب وخیم بیماری را گرفت. روش های درمان شامل جایگزینی پروتئین های رژیم غذایی با مخلوطی از اسید های آمینه فاقد لوسین، ایزولوسین و والین می باشد. هنگامی که سطح پلاسمایی این اسید های آمینه به حد طبیعی باز می گردد، مجدداً مقداری از این ترکیبات به شیر و سایر مواد غذایی اضافه می شود به نحوی که هیچ گاه از میزان مورد نیاز متابولیک شیر خوار فراتر نرود .
دیالیز به تنهایی روش مرجعی برای کاهش مقادیر بالای اسیدهای آمینه شاخه دار (BCAAs) نمی باشد مگر در موارد اورژانس که سیستم عصبی طفل به خطر افتاده باشد. یک درمان موثرتر، تجویز سرم های حاوی اسیدهای آمینه و عاری از BCAAs است که همراه با گلوکز وارد بدن می شود.پس از تزریق محلول که بطور اختصاصی بدین منظور تجویز می شود، لوسین، والین و ایزولوسین جهت سنتز پروتئینی در بدن مصرف می شوند (بدین ترتیب میزان این سه اسید آمینه شاخه دار BCAAs کاهش می یابد). در بعضی مواقع انسولین یا ترکیباتی مشابه، جهت سرعت بخشیدن به مصرف BCAAs نیز بطور جداگانه تجویز می گردد.تجویز مکمل حاوی ایزولوسین و والین جهت کمک به کمبودهای این دو اسید آمینه هنگامی که سطح سرمی آنها نسبت به لوسین بسرعت کاهش یافته، کمک کننده است، زیرا سطوح بسیار پایین ایزولوسین و والین می تواند سبب بروز جوشهای شدید پوستی در کودکان شود.
درمان دراز مدت MSUD مشتمل بر کنترل دائمی دقیق و محدودیت شدید رژیمی در مصرف پروتئین به منظور پیشگیری از تجمع BCAAs در خون است. ترکیب اصلی رژیم غذایی (کم پروتئین، دارای کربوهیدرات ) همراه با مصرف شیر خشک مخصوص کودکان مبتلا به MSUD است. مانند شیرخشک های Enfamil و Similac
این شیر خشک مخصوص حاوی کلیه ویتامین ها، مواد معدنی، و آمینو اسیدهای دیگر مورد نیاز برای تامین کالری و رشد کودک هستند. در عین حال محدودیت در مصرف این سه اسید آمینه نیزدر آن لحاظ شده است. تجویز تیامین و کارنیتین هم کمک کننده است.
پس از مصرف این شیرخشک، کنترل شدید سطوح خونی اسیدهای آمینه و انجام آزمایشات دیگر جهت تعدیل احتمالی ترکیب شیرخشک ضرورت دارد به نحوی که هر سه اسید آمینه شاخه دار BCAAs در بدن کودک در حد متعادل نگه داشته شوند. کودک گرفتار MSUD در هنگام رشد باید همچنان به مصرف شیرخشک مخصوص خود ادامه دهد و مقادیر توزین شده جیره غذایی بر حسب میزان لوسین روزانه تجویز شود.
شایان ذکر است که رژیم غذایی MSUD نمی تواند شامل غذاهای پرپروتئین از قبیل، گوشت، غلات، تخم مرغ و به مقدار کمتر محصولات لبنی، دانه ها (بادام زمینی) باشد. مصرف بیشتر میوه ها و سبزیجات زیر نظر پزشک بلامانع است. بعلاوه جهت پیشگیری از بروز بحران بیماری باید در مواقع ناخوشی، به کودک کربوهیدرات و مایعات به میزان کافی رسانده شود.
پیوند کبد: نیز از راههای درمان به شمار می رود (آنزیم مسوول تجزیه اسیدهای آمینه شاخه دار در کبد ساخته می شود، که با پیوند کبد این مشکل برطرف می گردد).
پیش آگهی
پیش آگهی بیماری تنها زمانی امیدوار کننده است که نقص مربوطه در نوزاد تا قبل از 5 روزگی تشخیص داده و درمان آغاز شود. ولی متأسفانه درهنگام انجام تست های غربالگری، اکثر نوزادان هنوزعلائم بیماری را از خود بروز نداده اند. بعلاوه روشهای مورد استفاده در بیشتر مراکز نیز قدیمی بوده و محاسبات ناصحیح دارند. بنابراین اغلب نوزادان تنها پس از شروع علائم بیماری تحت پیگیری قرار می گیرند.
پیش آگهی بیماری را می توان با توجه به جواب آنالیز اسیدهای آمینه BCAAsو تست خونی که نشان دهنده سطوح 20 اسید آمینه و ارتباط آنها با یکدیگر است، مشخص نمود.
در کل درمان سریع نوزاد (که باید تا آخر عمر ادامه یابد) در بیشتر موارد با رشد طبیعی همراه بوده، می تواند مانع از عقب ماندگی ذهنی وی گردد. با اینحال به دلایلی نامعلوم احتمال ابتلای این کودکان به بیش فعالی (ADHD)، اضطراب و افسردگی بیشتر از سایر کودکان است. بعضی کودکان حتی در صورت درمان به موقع، بعداً دچار ورم مغز و یا بحران متابولیک می شوند (در کودکانی که چندین بار دچار این بحران را تجربه کرده اند، احتمال آسیب مغزی و عقب ماندگی ذهنی بیشتر است).
متابولیسم فنیل آلانین و تیروزین
فنیل آلانین و تیروزین در یک مسیر بیوشیمیایی مشترک قراردارند که در اثر نقص در این چرخه حالاتی همانند موارد زیر ممکن است رخ دهد:
- فنیل کتونوری کلاسیک
فنیل کتونوری یک اختلال وراثتی بوده که موجب افزایش سطوح ماده ای به نام فنیل آلانین در خون می شود. فنیل آلانین جزء واحد های سازنده پروتئین ها(یا همان آمینو اسید) می باشد که از طریق رژیم غذایی به دست می آید. این آمینو اسید در همه پروتئین ها و برخی شیرین کننده های مصنوعی یافت می شود. اگر این بیماری درمان نشود، میزان بالای فنیل آلانین تا حدی افزایش می یابد که موجب ناتوانی ذهنی و سایر مشکلات شدید سلامتی می شود.
علائم و نشانه های این بیماری ممکن است ضعیف تا شدید باشند. شدیدترین فرم این بیماری فنیل کتونوری کلاسیک نامیده می شود. نوزادان مبتلا به فنیل کتونوری کلاسیک در ماه های اول زندگی خود، طبیعی به نظر می رسند. در صورتی که این نوزادان، درمان نشوند دچار عقب ماندگی ذهنی دائمی می شوند. تشنج، تکوین تاخیر یافته، مشکلات رفتاری و اختلالات روانی نیز در این بیماری شایع می باشد. افرادی که تحت درمان قرار نمی گیرند به دلیل افزایش فنیل آلانین در بدن ممکن است بوی موش یا کپک بدهند. کودکان مبتلا به فنیل کتونوری کلاسیک معمولاً دارای پوست و موی بور و روشن تر نسبت به سایر افراد خانواده می باشند و ممکن است اختلالات پوستی مانند اگزما داشته باشند.
اشکال ملایم تر این بیماری، گاهی اوقات واریانت فنیل کتونوری و هایپرآلانینمی غیر فنیل کتونوری نامیده می شوند که در این افراد خطر آسیب مغزی کمتر است. افرادی که به طور بسیار خفیف به این بیماری مبتلا می شوند نیازمند درمان با رژیم غذایی با میزان محدود فنیل آلانین نمی باشند.
نوزادان متولد شده از مادران مبتلا به فنیل کتونوری و دارای سطوح کنترل نشده ی فنیل آلانین (زنان فاقد رژیم غذایی با میزان محدود فنیل آلانین) در معرض خطر بالایی برای ابتلا به عقب ماندگی ذهنی هستند. زیرا این نوزادان قبل از تولد در رحم مادر مبتلای خود که رژیم با میزان کم فنیل آلانین مصرف نموده است، در معرض سطوح بالای فنیل آلانین قرار می گیرند. همچنین این نوزادان ممکن است دارای وزن پائینی در زمان تولد باشند و نسبت به سایر کودکان آهسته تر رشد نمایند. سایر مشکلات پزشکی مرتبط با این بیماری شامل نقایص قلبی یا سایر مشکلات قلبی، اندازه ی بسیار کوچک سر (میکروسفالی) و مشکلات رفتاری می باشند. هم چنین زنان مبتلا به فنیل کتونوری و سطوح کنترل نشده فنیل آلانین در معرض خطر بالایی برای سقط جنین می باشند.
اساس ژنتیکی
جهش در ژن PAH موجب ابتلا به بیماری فنیل کتونوری می شود. ژن PAH تولید کننده ی آنزیمی به نام فنیل آلانین هیدروکسیلاز می باشد. این آنزیم، آمینو اسید فنیل آلانین را به سایر ترکیبات مهم در بدن تبدیل می کند. اگر جهش های ژنی، فعالیت فنیل آلانین هیدروکسیلاز را کاهش دهد، فنیل آلانین موجود در رژیم غذایی به طور موثری متابولیزه نمی شود. در نتیجه این آمینو اسید در خون و سایر بافت ها به سطوح سمی می رسد . چون سلول های عصبی در مغز به سطوح فنیل آلانین حساس هستند، افزایش مقادیر این ماده می تواند موجب آسیب مغزی بشود.
فنیل کتونوری کلاسیک که شدیدترین فرم فنیل کتونوری می باشد زمانی رخ می دهد که فعالیت فنیل آلانین هیدروکسیلاز شدیداً کاهش یافته یا اصلا وجود ندارد. افراد مبتلا به فنیل کتونوری کلاسیک که تحت درمان قرار نمی گیرند دارای سطوح بالای فنیل آلانین می باشند که منجر به آسیب مغزی شدید و سایر مشکلات مرتبط به سلامت می شود. جهش در ژن PAH اگر طوری باشد که امکان فعالیت نسبی آنزیم را فراهم نماید موجب اشکال خفیف تر بیماری مثل واریانت PKU یا هایپرفنیل آلانینمی غیر فنیل کتونوری می شود.
تغییر در سایر ژن ها نیز ممکن است بر روی شدت فنیل کتونوری تاثیر داشته باشد. اما اطلاعات کمی در رابطه با این فاکتورهای ژنتیکی در دسترس می باشد.
فراوانی
شیوع فنیل کتونوری بین گروه های نژادی و مناطق جغرافیایی مختلف متفاوت است به طوری که شیوع آن در ایالات متحده ۱ در هر ۱۰ تا ۱۵ هزار نوزاد می باشد. بیشتر موارد فنیل کتونوری اندکی پس از تولد توسط غربالگری نوزادان شناسایی شده و سریعاً درمان شروع می شود. در نتیجه، علائم و نشانه های شدید فنیل کتونوری کلاسیک به ندرت مشاهده می شود.
الگوی توارث
این بیماری دارای الگوی توارث اتوزومی مغلوب می باشد.
اسامی دیگر بیماری
PKU
نقص PAH
بیماری فولینگ
بیماری نقص فنیل آلانین هیدروکسیلاز
- فنیل کتونوری غیر کلاسیک
فنیل کتونوری غیر کلاسیک یا نقص4HB
این بیماری متابولیکی در اثر نقص تولید 4HB اتفاق می افتد. شایعترین فرم آن کمبود PTPS است که شصت درصد موارد را در برمیگیرد و نقص DHPR در سه درصد موارد و در چهار درصد موارد نقص GTPCH را شامل می شود.
در آزمایشهای غربالگری نوزادی فنیل آلانین بالاست و سپس تستهای تکمیلی درخواست می شود.
علائم بالینی بیماری عبارتند از تشنج، حرکات غیر طبیعی و اختلال بلع و اختلال تون عضلانی(هیپوتونی) می باشد..
برای افتراق اختلالات متابولیسم HB۴ از سایر موارد هیپر فنیل آلانینمی، لازم است قبل از شروع رژیم با محدودیت فنیل آلانین، آزمایشهای مربوطه انجام شود..
در این موارد پترینهای ادرار شامل نئوپترین و بیوپترین اندازه گیری می شود و فعالیت آنزیم DHPR در خون نیز اندازه گیری می شود.
در صورتی که فعالیت این آنزیم نرمال باشد و فرد همچنان مشکوک به بیماری باشد و آزمایشهای نئوپترین و بیوپترین ادرار نرمال باشد احتمال نقص موارد شایع HB۴ کمتر مطرح میشود.
- آلکاپتونوری
آلکاپتونوری (Alkaptonuria) یا بیماری ادرار سیاه یک اختلال ارثی بسیار نادر است که از تجزیه کامل دو واحد پروتئینی (اسیدهای آمینه) به نام های تیروزین (tyrosine) و فنیل آلانین (phenylalanine) توسط بدن جلوگیری می کند و منجر به تجمع یک ماده شیمیایی به نام اسید هموژنتیسیک (homogentisic acid) در بدن می شود. تجمع این ماده شیمیایی می تواند ادرار و قسمت هایی از بدن را به رنگ تیره تبدیل کند و در طول زمان منجر به بروز طیف وسیعی از مشکلات سلامتی و عوارض جانبی شود. Ochronosis یا تجمع رنگدانه های تیره در بافت های همبند مانند غضروف و پوست نیز مشخصه این اختلال است و این رنگدانه آبی مایل به سیاه معمولاً بعد از 30 سالگی ظاهر می شود. افراد مبتلا به آلکاپتونوریا معمولاً در اوایل بزرگسالی به آرتریت به ویژه در ستون فقرات و مفاصل بزرگ مبتلا می شوند. از دیگر ویژگی های این بیماری می توان به مشکلات قلبی، سنگ کلیه و سنگ پروستات اشاره کرد. تا انتها با سیتوژنوم همراه باشید تا اطلاعات کاملی در رابطه با این بیماری متابولیک کسب کنید.
آلکاپتونوری که به آن اختصارا AKU نیز گفته می شود، یک اختلال متابولیک ژنتیکی نادر است و زمانی اتفاق می افتد که بدن انسان نتواند به اندازه کافی آنزیمی به نام دی اکسیژناز هموژنتیزیک (HGD) تولید کند؛ افراد مبتلا فاقد مقدار عملکردی کافی از این آنزیم برای تجزیه هموژنتیزیک اسید هستند که در نهایت با تجمع اسید هموژنتیزیک در بدنشان مواجه می شوند. تجمع اسید هموژنتیسیک باعث تغییر رنگ و شکنندگی استخوان ها و غضروف افراد مبتلا می شود و این امر معمولاً منجر به آرتروز به ویژه در ستون فقرات و مفاصل بزرگ می گردد. افراد مبتلا به آلکاپتونوریا ادراری دارند که وقتی در معرض هوا قرار می گیرد، قهوه ای تیره یا سیاه می شود. البته این تغییر رنگ ممکن است تا چند ساعت پس از ادرار رخ ندهد و اغلب مورد توجه قرار نمی گیرد.
آلکاپتونوری در اثر جهش در ژن هموژنتیز 1،2-دیاکسیژناز (HGD) ایجاد می شود. این ژن حاوی دستورالعمل هایی برای ایجاد (رمزگذاری) آنزیمی است که هموژنت 1،2-دیاکسیژناز نام دارد و برای تجزیه اسید هموژنتیزیک ضروری است. جهشهای ژن HGD منجر به کاهش مقدار آنزیم HGD می شود که به نوبه خود منجر به افزایش هموجنتیسیک اسید می گردد. اگرچه این محصول به سرعت توسط کلیه ها از بدن پاک می شود، اما به آرامی در بافت های مختلف بدن به ویژه بافت همبند مانند غضروف تجمع می یابد. با گذشت زمان (به ندرت قبل از بزرگسالی)، در نهایت رنگ بافت آسیب دیده را به آبی یا سیاه تغییر می دهد. تجمع طولانی مدت و مزمن اسید هموژنتیزیک در نهایت باعث ضعیف شدن و آسیب به بافت آسیب دیده می شود و منجر به بسیاری از علائم مشخصه آلکاپتونوری می گردد.
علائم بیماری آلکاپتونوری
علائم بیماری آلکاپتونوری از بدو تولد وجود دارد با این حال، علائم بیشتر و چشمگیرتر معمولاً تا بزرگسالی ظاهر نمی شوند. این علائم معمولاً به آرامی پیشرونده هستند و با افزایش سن شدت می یابند. از جمله علائم شایع این بیماری می توان به موارد زیر اشاره کرد:
ادرار افراد مبتلا به آلکاپتونوریا ممکن است به طور غیرطبیعی تیره باشد یا ممکن است با قرار گرفتن طولانی مدت در مجاورت هوا سیاه شود، با این حال از آنجایی که این تغییر اغلب چندین ساعت طول می کشد، اغلب مورد توجه قرار نمی گیرد. در دوران شیرخوارگی، پوشک ممکن است سیاه رنگ شود (در اثر قرار گرفتن در معرض ادرار در هوا)، اگرچه این مورد اغلب نادیده گرفته می شود.
در طی سالیان متمادی، اسید هموژنتیزیک به آرامی در بافت های سراسر بدن تجمع می یابد و تقریباً در هر ناحیه ای از بدن، از جمله غضروف، تاندون ها، استخوان ها، ناخن ها، گوش ها و قلب می تواند جمع شود. این تجمع، بافت ها را تیره می کند و باعث ایجاد طیف وسیعی از مشکلات می شود.
مشکلات مفاصل و استخوان ها: هنگامی که یک فرد مبتلا به آلکاپتونوریا به 30 سالگی می رسد، ممکن است مشکلات مفصلی را تجربه کند که به طور معمول، کمردرد و سفتی و به دنبال آن درد زانو، لگن و شانه خواهد بود؛ این ها علائم اولیه آرتروز هستند. در نهایت، غضروف هایش ممکن است شکننده شده و بشکند و منجر به آسیب مفاصل و ستون فقرات شود که ممکن است جراحی تعویض مفصل برای درمانش مورد نیاز باشد. در حالت کلی افراد مبتلا ممکن است دچار ناهنجاری هایی شوند که بر تاندون ها تأثیر می گذارد، از جمله تاندون های آشیل ضخیم شده و التهاب تاندون ها (تاندونیت). تاندون ها و رباط های آسیب دیده ممکن است در معرض پارگی قرار بگیرند.
عوارض چشم و گوش: بسیاری از افراد مبتلا روی سفیدی چشم خود لکه های قهوه ای یا خاکستری مشاهده خواهند کرد. علامت دیگر در بسیاری از بزرگسالان مبتلا به آلکاپتونوریا ضخیم شدن غضروف گوش است که همچنین ممکن است آبی، خاکستری یا سیاه به نظر برسد که به آن اکرونوز می گویند. جرم گوش مبتلایان نیز ممکن است سیاه یا قهوه ای مایل به قرمز باشد.
عوارض پوست و ناخن: آلکاپتونوریا می تواند منجر به تغییر رنگ عرق شود که می تواند لباس ها را لکه دار کند و باعث شود که برخی از افراد دارای نواحی آبی یا سیاه روی پوست خود شوند. رنگ ناخن ها نیز ممکن است مایل به آبی یا قهوه ای شود. تغییرات رنگ پوست نواحی در معرض نور خورشید و جایی که غدد عرق یافت می شوند (مانند گونه ها، پیشانی، زیر بغل و ناحیه تناسلی) آشکارتر است.
مشکلات تنفسی: اگر استخوان ها و ماهیچه های اطراف ریه ها سفت شوند، می توانند از انبساط قفسه سینه جلوگیری کنند و منجر به تنگی نفس یا مشکل در تنفس شوند.
مشکلات قلبی، کلیه و پروستات: رسوبات اسید هموژنتیزیک در اطراف دریچه های قلب می تواند باعث سفت شدن و شکننده و سیاه شدن آن ها شود؛ رگ های خونی نیز می توانند سفت و ضعیف شوند. این عارضه می تواند منجر به ایجاد بیماری قلبی شود و ممکن است فرد را نیازمند تعویض دریچه قلب نماید. این رسوب همچنین می تواند منجر به سنگ کلیه، سنگ مثانه و سنگ پروستات شود.
توارث آلکاپتونوریا
هر سلولی که در بدن انسان های سالم وجود دارد (فارغ از جنسیتشان) شامل 23 جفت کروموزوم (46 عدد) است؛ این کروموزوم ها حامل ژن هایی هستند که افراد از والدین خود به ارث برده اند. یکی از هر جفت کروموزوم از هر یک از والدین به ارث می رسد (به استثنای کروموزوم های جنسی)، به این معنی که دو نسخه از هر ژن در هر سلول وجود دارد. ژن دخیل در ایجاد بیماری آلکاپتونوری ژن HGD نام دارد. این ژن دستورالعمل لازم برای ساخت آنزیمی به نام هموژنتیزات اکسیداز (که برای تجزیه اسید هموژنتیزیک لازم است) را ارائه می دهد. الگوی توارث این بیماری اتوزوم مغلوب است یعنی برای ایجاد آلکاپتونوریا باید دو نسخه از ژن معیوب HGD (یک نسخه از هر والدین) را به ارث ببرید. احتمال بروز این بیماری بسیار اندک است (به همین دلیل است که این وضعیت بسیار نادر می باشد) با این حال در کودکان حاصل از ازدواج های فامیلی شایع تر است. والدین یک فرد مبتلا به آلکاپتونوریا اغلب فقط یک نسخه از ژن معیوب را دارند، به این معنی که هیچ علامت یا نشانه ای از این بیماری را ندارند اما ناقل آن به حساب می آیند. خطر این که دو والد ناقل، هر دو ژن معیوب را به فرزند خود منتقل کنند و در نتیجه فرزندی مبتلا داشته باشند در هر بارداری 25 درصد است. خطر داشتن فرزندی که مانند والدین ناقل است در هر بارداری 50 درصد است. شانس اینکه کودک از هر دو والدین ژن طبیعی دریافت کند و از نظر ژنتیکی برای آن صفت خاص طبیعی باشد 25 درصد است. لازم به ذکر است که این خطر برای مونث و مذکر یکسان می باشد و ربطی به جنسیتشان ندارد.
تشخیص آلکاپتونوریا
پزشک عمومی، پزشک متخصص داخلی و یا اورولوژیستی که تحت نظرش هستید ممکن است با شنیدن تغییر رنگ ادرار و مدفوع شما به وجود بیماری آلکاپتونوری مشکوک شود. وی ممکن است برای رسیدن به تشخیص دقیق و سریع تر از تکنیک های زیر کمک بگیرد:
آزمایش ادراری: مقادیر زیاد هموژنتیزیک اسید در ادرار را می توان با آنالیز کروماتوگرافی گازی – طیف سنجی جرمی تشخیص داد.
تصویربرداری پزشکی: برای تعیین وجود و میزان بیماری مفصلی و نخاعی یا درگیری دریچه های آئورت یا میترال می توان از تکنیک های مختلف تصویربرداری مانند سی تی اسکن و یا MRI استفاده کرد.
آزمایش ژنتیک مولکولی: این تست می تواند جهش در ژن HGD را تشخیص دهد با این حال، انجام این آزمایش برای تأیید تشخیص الزامی نیست.
اکوکاردیوگرافی: در افراد بالای 40 سال، اکوکاردیوگرافی ممکن است برای تشخیص قوی توصیه شود. با اکوکاردیوگرافی، امواج صوتی از قلب منعکس میشوند (پژواک)، که پزشکان را قادر می سازد تا عملکرد و حرکت قلب را بررسی کنند.
آلکاپتونوری یک بیماری مادام العمر است و در حال حاضر هیچ درمان خاص و قطعی برای آن وجود ندارد. با این حال برای کنترل عوارض و علائمی که ایجاد می کند راهکارهای زیر پیشنهاد می شوند:
دارو درمانی: افراد مبتلا به آلکاپتونوریا اغلب داروهای ضد التهابی برای درمان درد مفاصل دریافت می کنند و در موارد شدید، ممکن است داروهای قوی تری مانند مواد مخدر برایشان تجویز شود. مدیریت و کنترل درد متناسب با مورد خاص هر فرد تنظیم می شود و نیاز به پیگیری و تنظیم طولانی مدت دارد.
درمان های حمایتی: برخی از افراد مبتلا به آلکاپتونوریا از فیزیوتراپی و کاردرمانی بهره مند می شوند که می تواند به حفظ قدرت و انعطاف پذیری عضلات و مفاصلشان کمک کند.
عمل جراحی: برخی از افراد مبتلا به آلکاپتونوریا نیاز به مداخله جراحی دارند به طوریکه تقریباً نیمی از آن ها نیاز به تعویض مفصل ران، زانو یا کتف در سنین 50 تا 60 سالگی خواهند داشت. همچنین به ندرت پیش می آید که این افراد به جراحی ستون فقرات، از جمله فیوژن و یا برداشتن دیسک ها نیاز داشته باشند. جراحی برای تعویض دریچه آئورت یا میترال نیز ممکن است ضروری باشد.
مصرف ویتامین سی: در کودکان و بزرگسالان، دوزهای بالای ویتامین C نیز برای درمان آلکاپتونوریا استفاده شده است، زیرا مانع از تجمع و رسوب اسید هموژنتیزیک می شود. با این حال مؤسسه ملی سلامت امریکا هشدار می دهد که استفاده طولانی مدت از ویتامین C گاهی اوقات می تواند تولید سنگ کلیه را افزایش دهد و به طور کلی برای درمان طولانی مدت این بیماری بی اثر است.
داروی نیتیسنون: محققان در حال مطالعه استفاده از دارویی به نام نیتیزینون یا nitisinone (Orfadin®) به عنوان یک درمان بالقوه برای آلکاپتونوری هستند. این دارو که در سال 2001 تاییدیه خود را از سازمان غذا و دارو (FDA) دریافت کرد، برای درمان یک اختلال متابولیک به نام تیروزینمی تایید شده بود. در مطالعات قبلی نشان داده شده بود که نیتیسنون به طور قابل توجهی تجمع اسید هموژنتیزیک را در افراد مبتلا به آلکاپتونوری کاهش می دهد. با این حال، تحقیقات بیشتری برای تعیین اینکه آیا تجویز نیتیزینون برای بیماران جوانتر می تواند از علائم آلکاپتونوری جلوگیری کند یا نه و همچنین برای تعیین ایمنی و اثربخشی طولانی مدت دارو برای افراد مبتلا به AKU ضروری است.
- تیروزینمی
یماری تیروزینمی (Tyrosinemia) یک ناهنجاری متابولیک مادرزادی است که در آن بدن نمیتواند به نحو مؤثر ی اسید آمینه تیروزین را تجزیه نماید. تیروزین از طریق مواد غذایی وارد بدن می شود یا در داخل بدن از فنیل آلانین ساخته می شود. تیروزین برای سنتز پروتئین و به عنوان پیش ساز برای ساخته شدن دوپامین، نوراپی نفرین، اپی نفرین، ملانین و تیروکسین به کار می رود. نشانههای این بیماری شامل اختلالات کبد و کلیه و نیز عقب ماندگی ذهنی بوده و در صورت عدم درمان، میتواند موجب مرگ بیمار گردد. بیشتر انواع تیروزینمی باعث هایپرتیروزینمی (افزایش سطح تیروزین در خون) میشوند.
هایپر تیروزینمی اکتسابی ممکن است در بیماریهای هپاتوسلولار بسیار شدید (نارسایی کبد)، کمبود ویتامین C و هایپرتیروئیدیسم دیده شود. هایپرتیروزینمی در نوزادان نارس در صورت نمونه گیری خیلی سریع بعد از تغذیه، دیده می شود.
انواع تیروزینمی
تیروزینمی دارای سه نوع مختلف است که هر یک دارای نشانههای خاص خود میباشد:
- تیروزینمی نوع I: نوع کبدی کلیوی (Hepatorenal form)، که ناشی از نقص در آنزیم فوماریل استو استات هیدرولاز می باشد.
- تیروزینمی نوع II: نوع چشمی پوستی که سندرم ریچنر-هانهارت (Richner-Hanhart syndrome) نیز خوانده میشود، نوعی ناهنجاری مادرزادی متابولیسم و ناشی از نقص در آنزیم تیروزین آمینو ترانسفراز (در سیتوزول) می باشد.
- تیروزینمی نوع III: نوعی ناهنجاری متابولیک نادر است که به دنبال نقص در آنزیم ۴- هیدروکسی فنیل پیروات دی اکسیژناز ایجاد می شود. این آنزیم که به طور عمده در کبد (و به میزان کمتری در کلیه) یافت میشود، یکی از آنزیمهای مورد نیاز برای تجزیه تیروزین است (این آنزیم یکی از فرآورده های جانبی تیروزین به نام ۴-هیدروکسی فنیل پیروات را به اسید هموجنتیسیک تبدیل میکند). از ویژگیهای مشخص تیروزینمی نوع سه میتوان به عقب ماندگی ذهنی، تشنج و دورههای از دست دادن دوره ای تعادل (آتاکسی متناوب) اشاره کرد.
متابولیسم:
1- ابتدا تیروزین در کبد تحت اثر آنزیم تیروزین آمینوترانسفراز (یا تیروزین ترانس آمیناز) به 4-هیدروکسی فنیل پیرووات تبدیل می شود (نقص در این آنزیم منجر به تیروزینمی تیپ II می گردد.
2- سپس 4-هیدروکسی فنیل پیرووات تحت تاثیر 4-هیدروکسی فنیل پیرووات دی اکسیژناز (p-hydroxy phenylpyrovate dioxigenase) به homogentsate تبدیل می شود (نقص در این آنزیم منجر به تیروزینمی تیپ III می گردد.
3- آنگاه Homogentsate تحت تاثیر homogentsate oxidase به 4-maleylacetoacetate تبدیل می شود نقص در این آنزیم منجر به ایجاد بیماری alcaptonuria می گردد.
4- در ادامه 4-maleylacetoacetate تحت تاثیر 4-maleylacetoacetate isomerase به 4- fumarylacetoacetate می شود.
5- و نهایتاً 4-fumarylacetoacetate تحت تاثیر 4-fumarylacetoacetate acetase به فومارات و استواستات تبدیل می شود (نقص در این آنزیم منجر به تیروزینمی تیپ I می گردد).
نقش توارث در بیماری تیروزینمی
این نقص معمولاً از الگوی وراثتی اتوزومال مغلوب پیروی میکند. در این الگو بیماران مبتلا باید دو کپی (= نسخه) از ژن جهش یافته داشته باشند تا علائم بیماری در آنها بروز کند. افرادی که دارای یک ژن معیوب هستند، ناقل خوانده میشوند و علائم بیماری را نشان نمیدهند، اما میتوانند ژن معیوب را به فرزندان خود منتقل کنند. وقتی هر دو والد ناقل باشند، 25% احتمال تولد کودک سالم، 50% احتمال تولد کودک ناقل و 25% احتمال تولد کودک مبتلا به تیروزینمی در هر بارداری برای آنها وجود دارد.
شیوع تیروزینمی
شیوع تیروزینمی تیپ I در اسکاندیناوی یک مورد در هر صد هزار تولد زنده و در شمال شرقی ایالت کبک کانادا (کانادایی های فرانسوی تبار) 1.46 مورد در هر هزار تولد زنده است. این بیماری در تمامی نژادهای قومی موجود در سرتاسر جهان مشاهده شده است (اصطلاحا یک بیماری Panethnic است). با این حال پیش بینی می شود، شیوع آن در کشورهایی که ازدواجهای فامیلی رواج بیشتری دارد (مثل ایران بیشتر باشد ( تیروزینمی تیپ II در ایتالیا شایع است.
علائم بالینی تیروزینمی
تیروزینمی تیپ I
آسیب ارگانی در اثر تجمع متابولیت های حاصل از شکسته شدن تیروزین بخصوص سوکسینیل استن ایجاد می شود. علایم این بیماری در افراد مختلف، متفاوت است. بیماری تیروزینمی نوع 1 به دو شکل حاد و مزمن دیده می شود. فرم متداول آن در نوزادان شیوع بیشتری دارد و نوع دیگر آن در کودکان و بزرگسالان بیشتر مشاهده می شود.
تیروزینمی نوع 1 در نوزادان (فرم حاد)
کودکان مبتلا به این اختلال، در صورت عدم درمان، شیرخواران مبتلا در بدو تولد نرمال هستند اولین علایم بیماری را در چند ماه اول زندگی خود (6-2 ماهگی) نشان می دهند. این بیماران بندرت قبل از یک ماهگی ویا بعد از سال اول زندگی علامت دار می شوند. هرچه تظاهر بیماری زودتر باشد پیش آگهی بیمار بدتر می باشد. تعدادی از این علایم عبارتند از:
اسهال و مدفوع خونی
کاهش قند خون
ضعف و خواب آلودگی مفرط
استفراغ
زودرنجی، حساسیت و تحریک پذیری
عدم وزن گیری مناسب
استشمام بویی شبیه بوی کلم پخته در پوست و ادرار کودک (بعلت افزایش متابولیت های متیونین)
مشکلات کبدی در این بیماران متداول بوده و بصورت یک بحران حاد کبدی در شروع بیماری دیده می شود، بطوریکه می تواند باعث عوارض زیر شود:
بزرگی کبد
نارسایی عملکرد کبدی (بالا رفتن سطح سرمی ترانس آمینازها)
زردی پوست
خون ریزی و کبودی
ورم پاها و شکم
همچنین مشکلات کلیوی نیز در این بیماران متداول بوده و می تواند باعث عوارض زیر گردد:
آسیب توبولی کلیه به صورت سندرم فانکونی،
اسیدوز متابولیک با آنیون گپ نرمال
هیپرفسفاتوری و هیپوفسفاتمی،
راشتیسم مقاوم به درمان
بزرگی کلیه
نفروکلسینوز
نرمی استخوان
اگر درمان سریع و مناسب در این بیماران انجام نشود، اغلب کودکان مبتلا، به علت مشکلات کبدی و کلیوی می میرند.
در تعدادی از کودکان علایم زیر مشاهده می شود:
درد و ضعف (به خصوص در ناحیه پاها)
تاخیر در راه رفتن
مشکلات تنفسی
تپش قلب
تشنج
وضعیت هیپر اکستانسیون شدید سر و تنه،
کوما (که گاهی منجر به مرگ بیمار می شود)
تیروزینمی نوع I در کودکان (فرم مزمن):
کودکانی که مبتلا به فرم مزمن این بیماری هستند، معمولاً اولین علایم را بعد از دو ماهگی نشان می دهند. اولین نشانه های بیماری عبارتند از: عدم وزن گیری مناسب، بوی خاصی شبیه بوی کلم پخته، اسهال و استفراغ متناوب، کاهش قند خون. با گذشت زمان، مشکلات کلیوی، کبدی و عصبی نیز در این افراد مشاهده می شود. این کودکان هوش نرمالی دارند.
کبد: در صورت عدم درمان، برخی بیماریهای کبدی (نظیر بزرگی کبد و سیروز کبدی) ایجاد می شود که می توانند منجر به نارسایی کبد گردند. در کودکان درمان نشده، نارسایی یا سرطان کبد تا قبل از سن 10 سالگی مشاهده می شود. اگر درمان سریعا آغاز شود می تواند از ایجاد نارسایی کبد در بسیاری از کودکان جلوگیری کند.
کلیه ها: در کودکان درمان نشده، مشکلات حاد کلیوی (بخصوص آسیب توبولی کلیه به صورت سندرم فانکونی) دیده می شود. هنگامی که کلیه ها به طور طبیعی فعالیت نمی کنند دوره هایی از استفراغ، ضعف و تب در بیمار ایجاد می شود. نرمی استخوان در کودکانی که مشکلات کلیوی دارند نیز شایع است. اگر درمان سریعا آغاز شود می تواند از ایجاد مشکلات کلیوی در بسیاری از کودکان جلوگیری کند.
بحرانهای عصبی: تعدادی از کودکان از ضعف، درد و یا بی حسی دستها و پاها و دیگر اندامها (بصورت گزگز و مورمور شدن) رنج می برند. مشکلات تنفسی و تپش قلب نیز در گروهی از این بیماران مشاهده می شود. تعدادی از کودکان دچار تشنج می شوند که ممکن است باعث ایجاد کوما در آنها گردد. اگر درمان سریعا آغاز شود، می تواند از ایجاد بحرانهای عصبی در بسیاری از این کودکان جلوگیری کند.
دیگر علایم: در برخی از بیماران ضعف عضلانی که ممکن است به سمت فلج پیش رود، بزرگی طحال، واریس مری و خونریزی از آن، هیپرتروفی جزایر لوزالمعده (که بدون علامت است)، مشکلات قلبی (بصورت افزایش ضربان قلب) و افزایش فشار خون نیز مشاهده می شود.
تیروزینمی تیپ II:
زمان تولد: طبیعی
دوره نوزادی، شیرخوارگی و یا بزرگسالی:
تظاهرات چشمی: اشک ریزش، ترس از نور، درد چشمها یا سابقه قرمزی آنها، خراش، زخم و یا پلاک در قرنیه و کدورت قرنیه است که به آن Kerititis می گویند که ناشی از رسوب کریستالهای تیروزین در قرنیه است. تظاهرات چشمی زودتر از تظاهرات پوستی اتفاق می افتد و برخلاف ضایعات هرپسی دو طرفه است.
تظاهرات پوستی: کراتوزهای دردناک در نواحی تحت فشار کف دست و پاها (Palmoplantar hyperkeratosis)، ضایعات در نزدیک نوک انگشتان و در زیر ناخنها بصورت پاپول یا پلاک با حاشیه نامنظم ولی مشخص بوده که بصورت تو خالی و یا زخمی شونده و بدون خارش اما دردناک هستند و به صورت نواحی هیپرکراتوتیک بر روی زانو، آرنج، قوزک پا و یا زبان نیز دیده می شوند.
عوارض دیگر: بیش فعالی، تکامل غیرطبیعی در تکلم، ناتوانی یادگیری، خود آزاری و در نیمی از موارد عقب ماندگی ذهنی خفیف رخ می دهد.
در تیروزینمی تیپ III علائم بالینی نامعلوم است و ضایعات پوستی ندارد. تاخیر تکامل، تشنج، آتاکسی متناوب و رفتارهای خود تخریبی گزارش شده است.
طی غربالگری بیماریهای متابولیک در نوزادان 2 تا 7 روزه با استفاده از تست MS/MS مارکرهای مربوط به آسیل کارنیتن ها اندازه گیری می شود (از جمله مارکرهایی که طی این غربالگری اندازه گیری می شوند می توان از تیروزین نام برد)، که بعنوان مارکرهای اولیه غربالگری تیروزینمیا تیپ II و III محسوب می گردند.
در بعضی از پروتکل ها سطح سوکسینیل استن هم اندازه گیری می شود که به عنوان مارکر اولیه در تیروزینمیا تیپ I استفاده می شود و حساسیت و اختصاصیت بالاتری از تیروزین دارد.
در صورتی که درغربالگری نتیجه تست فوق بالاتر از حد نرمال باشد، باید تست MS/MS یک تا دو روز بعد با خونگیری مجدد تکرار شود. بدیهی است در صورت وجود مشکل آنزیمی، میزان این مارکرها در نمونه دوم افزایش بیشتری پیدا خواهند کرد.
مارکرهای ثانویه: بعد از تائید افزایش مارکرهای اولیه طی دو مرحله نمونه گیری فوق، تغییرات مارکرهای ثانویه طی دو مرحله نمونه گیری نیز با محاسبه نسبت های زیر چک می شود:
افزایش تیروزین و نسبت تیروزین به سیترولین در تیروزینمیا تیپ I.
افزایش نسبت تیروزین به سیترولین در تیروزینمیا تیپ II و III.
در صورت مشاهده افزایش نسبت های فوق و وجود این تغییرات در مارکرهای ثانویه، جواب فرد به عنوان یک جواب "خارج از محدوده نرمال" تلقی و به صورت "مشکوک" از نظر تیروزینمی گزارش شده و انجام تست های تائیدی یا تشخیصی برای تائید تشخیص بیماری توصیه می گردد.
تست های تشخیصی تیروزینمی
تشخیص تیروزینمی تیپ I:
- بررسی اسیدهای آلی ادرار: افزایش سوکسینیل استون بالا (که جنبه تشخیصی داشته و گاهی میتواند طبیعی باشد)، افزایش مشتقات ۴-هیدروکسی فنیل این ماده نظیر 4-Hydroxyphenylacetate، 4-Hydroxyphenyllactate، 4-Hydroxyphenylpyrovate،
- اسیدهای آمینه پلاسما: افزایش یا طبیعی بودن تیروزین، افزایش متیونین،
- پورفیرینهای ادرار: افزایش دلتا آمینو لوولینیک اسید (بدلیل مهار فعالیت آنزیم دلتا آمینو لوولینیک هیدراتاز توسط سوکسینیل استن)،
- آلفا فتوپروتئین سرم: طبیعی یا بالا،
- هیپرفسفاتوری و هیپوفسفاتمی،
- آزمونهای کبدی: غیر طبیعی ولی سطح بیلی روبین معمولأ نرمال است و فقط در نارسایی کبد بالا می رود،
- کاهش سطح فاکتورهای انعقادی تولید شده در کبد،
- سایر آزمونها: کاهش گلوکز خون،افزایش زمان پروترومبین و زمان نسبی ترومبوپلاستین، آمینواسیدوری ژنرالیزه (=عمومی)،
- تست های مولکولی: بررسی موتاسیون ژن FAH
تشخیص تیروزینمی تیپ II:
- بررسی اسیدهای آلی ادرار: افزایش ترکیباتی نظیر 4-Hydroxyphenylacetate، 4-Hydroxyphenyllactate، 4-Hydroxyphenylpyrovate علی رغم بلاک آنزیمی، لاکتات و استات بالاست. این فرضیه وجود دارد که در هنگام تیروزین بالا ترانس آمینازهای دیگر فعال می شوند و 4-هیدروکسی فنیل پیرووات در سایر ارگانل های داخل سلولی مثل میتوکندری تولید می شود،
- آزمونهای عملکرد کبد و کلیه: نرمال است برخلاف تیروزینمیای تیپ I،
- اسیدهای آمینه پلاسما: افزایش شدید تیروزین،
- آمینواسیدهای ادرار: افزایش تیروزین،
- آمینواسیدهای مایع مغزی نخاعی: افزایش تیروزین،
تشخیص تیروزینمی تیپ III:
- افزایش متوسط و دائم تیروزین در اندازه گیری اسیدهای آمینه به روش HPLC،
- بررسی فعالیت آنزیم 4-هیدروکسی فنیل پیروات دی اکسیژناز (HpPD) در نمونه بیوپس کبد،
- بررسی مولکولی موتاسیون آنزیم 4-هیدروکسی فنیل پیروات دی اکسیژناز (HPD).
تشخیص تیروزینمی قبل از تولد
در مورد تیپ I تشخیص بر مبنای بررسی آنزیم در آمنیوسیتهای کشت داده شده و یا بررسی مواد حاصل از نمونه گیری از پرزهای جفتی (= کوریونی) و یا اندازهگیری غلظت سوکسینیل استون در مایع آمنیوتیک صورت میگیرد. اگر موتاسیون خانوادگی مشخص شده باشد با آنالیز DNA در آمنیوسیت ها یا پرزهای کوریونی امکان پذیر می باشد. در مورد تیپ II و III تشخیص قبل از تولد وجود ندارد.
درمان تیروزینمی
برای تیروزینمی تیپ I داروی نیتیزینون (Nitisinone) که در واقع همان ۲- نیترو ۴- تری فلورومتیل بنزوئیل ۳-۱ سیکلو هگزان دیول میباشد به میزان ۱ تا ۲ میلی گرم به ازای هر کیلوگرم وزن بیمار، دو بار در روز تجویز می شود (این ماده مهار کننده ۴ هیدروکسی فنیل پیرووات دی اکسیژناز بوده و باعث توقف تجمع متابولیتهای سمی و بالا رفتن تیروزین میشود).
رعایت رژیم غذایی با محدودیت فنیل آلانین و تیروزین در هنگام مصرف این دارو ضروری است. در صورت بروز نارسایی کبد یا کارسینوم سلول کبدی پیوند کبد ضرورت می یابد.
سالها قبل که استفاده از داروی nitisinone متداول نبود، پیوند کبد یک روش درمانی مناسب برای بیماران مبتلا به تیروزینمی نوع I محسوب می شد. اکنون این دارو از بروز بسیاری از مشکلات کبدی در مبتلایان به این بیماری، جلوگیری می کند. برای بررسی اینکه آیا این دارو توانایی جلوگیری از سرطانهای کبدی را دارد یاخیر، به زمان بیشتری نیاز است. در بسیاری از بیماران داروی nitisinone، مشکلات کبدی را به تاخیر می اندازد و امید می رود که بتواند جایگزین پیوند کبد در این بیماران شود.
بیشترین تاثیر این دارو در بیمارانی وجود دارد که توسط غربالگری نوزادی تشخیص داده می شوند و قبل از اینکه علائم شروع شود درمان آغاز می شود. در تمامی سنین نیتیزینون باعث کاهش نیاز به پیوند کبد، کاهش حملات ناشی از نارسایی کبد و دردهای نورولوژیک می شود.
پیوند کبد، هنوز هم به عنوان راه حلی در جلوگیری از سرطان کبد مدنظر قرار می گیرد، بعلاوه کودکانی که اکنون علایم سرطان کبد یا نارسایی کبد را دارند نیز بکار می رود. این دارو برخی از آسیب های کبدی که قبل از درمان اتفاق افتاده است قابل برگشت نیستند.
جهت پیشگیری از نرمی و یا پوکی استخوان تجویز ویتامین D3 ضروری است.
رژیم غذایی تیروزین و فنیل آلانین کم
این رژیم، شامل مواد غذایی با سطح پایینی از فنیل آلانین و تیروزین می باشد. به عبارت دیگر کودک شما باید از شیر گاو، شیر خشکهای معمولی، گوشت، تخم مرغ و پنیر پرهیز کند. همچنین آرد معمولی، باقلا خشک شده و مغزها ( گردو و بادام و غیره) نیز دارای فنیل آلانین و تیروزین هستند و باید شدیداً محدود شوند. میوه ها و سبزیجات اغلب فنیل آلانین و تیروزین کمی دارند و در مقادیر تعیین شده، می توانند مورد استفاده قرار گیرند. همچنین مواد غذایی طبی خاصی وجود دارند که مناسب افراد مبتلا به تیروزینمی نوع I می باشند همانند: آرد های مخصوص، پوره های مخصوص، نان و برخی از ماکارونی ها. پزشکان و متخصصین تغذیه رژیم غذایی مناسب کودک شما را به مرور زمان طراحی خواهند کرد. برنامه غذایی کودک شما به چندین پارامتر وابسته است همانند: سن، وزن، حال عمومی و روند درمان کودک.
توجه شود این رژیم غذایی می تواند پیشرفت بیماری را آهسته کند ولی پیشرفت آنرا متوقف کند.
درمان در تیروزینمی تیپ II، شامل رژیم غذایی با محدودیت تیروزین و فنیل آلانین است. در زنان مبتلا کنترل رژیمی در طی حاملگی ضروری میباشد، زیرا عقب ماندگی ذهنی، کوچکی چشم و تشنج در کودکان مادران درمان نشده گزارش گردیده است.
درمان در تیروزینمی تیپ III هم شامل رژیم غذایی با محدودیت فنیل آلانین و تیروزین میباشد. همچنین درمان با ویتامین C بعنوان کوفاکتور 4-هیدروکسی فنیل پیروات دی اکسیژناز در نظر گرفته می شود.
تیروزینمیای گذرای نوزادی
در گروه کوچکی از شیرخواران تازه متولد شده ممکن است تیروزین بطور گذرا در 2-1 هفته اول زندگی بالا برود. بیشتر این نوزادان ناقص بوده و رژیم غذایی پرپروتئین دریافت می کنند. بعلت تاخیر در تکامل آنزیم 4-هیدروکسی فنیل پیروات دی اکسیژناز می باشد. شکایت اصلی این نوزادان بی حالی، ضعف درشیر خوردن و کاهش حرکات می باشد. بطور همزمان فنیل آلانین و تیروزین بالا داشته و سطح ادراری 4-هیدروکسی فنیل پیرویک اسید و متابولیت های آن در ادرار دیده می شود. معمولأ خود به خود درمان می شود و با کاهش پروتئین رژیم غذایی به کمتر از 2 گرم در هر کیلو وزن بدن به همراه تجویز ویتامین C توصیه می شود.
پیش آگهی تیروزینمی
در تیروزینمی تیپ I پیش آگهی با تجویز نیتیسیتون خوب است، اما در تیپ II امکان بروز عقب ماندگی ذهنی علیرغم درمان وجود دارد.
اغلب کودکان مبتلا به تیپ I بیماری که بعد از تولد سریعاً تحت درمان قرار گرفته اند، اکنون فاقد مشکلات حاد کبدی، کلیوی و عصبی می باشند. کودکان درمان شده، اغلب سطح هوشی و رشد نرمالی خواهند داشت. کودکانی که تا چند هفته بعد از تولد، تحت درمان قرار نگرفته اند، ممکن است مشکلات کبدی و کلیوی و عصبی داشته باشند. نرمی استخوان و تاخیر در رشد و نمو نیز ممکن است در این بیماران وجود داشته باشد. آثار تاخیر درمان در افراد مختلف، متفاوت است.
آیا امکان دارد که، دیگر اعضای خانواده مبتلا به تیروزینمی نوع I و یا ناقل آن باشند؟
امکان اینکه خواهر و برادران بزرگتر کودک مبتلا، که به طور طبیعی رشد کرده و به نظر سالم می رسند، به این بیماری مبتلا باشند کم است. با این وجود، جستجوی بیماری در دیگر اعضای خانواده بسیار مفید خواهد بود، زیرا از بسیاری از مشکلات جسمی پیشگیری خواهد کرد. از مشاور ژنتیک و یا پزشک متخصص سوال کنید که آیا دیگر اعضای خانواده باید آزمایشهای تشخیصی را انجام دهند یا خیر. برادران و خواهران دیگر بیمار که تیروزینمی نوع I ندارند ممکن است همانند والدین خود ناقل باشند. در ناقلین هیچ اختلالی مشاهده نمی شود. تستهای تشخیص ناقلین به جز در موارد خاص فقط در افراد بالای 18 سال قابل انجام است. برادران و خواهران هریک از والدین نیز 50% احتمال دارد که ناقل این بیماری باشند. اطلاع از ناقل بودن از این جهت اهمیت دارد که آنها نیز ممکن است بچه های مبتلا به این بیماری داشته باشند. اگر نتیجه آزمایش DNA برای تشخیص ناقلین، قانع کننده نبود، متدهای دیگری نیز وجود دارند که می توانند ناقلین را شناسایی کنند. هنگامی که هر دو والد می دانند که ناقل بیماری هستند و یا زمانی که یک کودک مبتلا به بیماری در خانواده وجود دارد، دیگر کودکانی که در این خانواده ها متولد می شوند باید علاوه بر آزمایشهای غربالگری تحت آزمایشهای دقیقتر، برای تشخیص تیروزینمی نوع 1 قرار گیرند.
نحوه پیگیری بیماران متابولیک بعد از تشخیص:
باید به والدین این گونه بیماران آموزش داده شود که نیاز به درمان مادام العمر بوده و رعایت نکات تغذیه ای (رژیم حاوی شیر خشک های مخصوص فاقد فنیل آلانین و تیروزین) در این بیماران برای برخورداری از یک روند رشد، سلامت و تکامل طبیعی اجباری می باشد.
این بیماران باید توسط یک متخصص بیماریهای متابولیک تحت مراقبت منظم قرار گیرند.
پیگیری درازمدت بیماری باید به والدین توصیه شود تا وضعیت نوزاد همیشه در حالت سالم قرار گیرد. مشورت با گروهی از مشاورین شامل متخصص اطفال، ژنتیک و تغذیه نیز باید صورت بگیرد و والدین باید بدانند که کوچکترین تخطی از رژیم غذایی می تواند مرگ فرزندشان را به همراه داشته باشد.
کودک شما به انجام تستهای مکرر خون و ادرار به منظور کنترل سطح اسیدهای آمینه، سوکسینیل استون، nitisinone، و کنترل عملکرد کبد و کلیه نیاز خواهد داشت. نتیجه این تستهای آزمایشگاهی رژیم غذایی و داروهای کودک شما را تعیین خواهند کرد.
برخی متخصصین، به بیماران خود پیشنهاد می کنند که سالی یک بار سی تی اسکن و MRI به منظور کنترل فعالیت کبد انجام دهند.
مشاوره ژنتیک برای تشخیص مولکولی بیماری و تعیین ریسک ابتلاء فرزند بعدی در صورت داشتن تصمیم به بارداری در آینده توصیه می شود.
در هر کودکی که دچار التهاب قرنیه میشود، غلظت تیروزین خون نیز باید اندازهگیری شود.
متابولیسم اسید های آمینه سولفور دار
در بیماری های این گروه می توان به اختلال هموسیستینوری اشاره کرد که الگوی توارث اتوزومی مغلوب داشته و با ناتوانی یادگیری، حملات عصبی ناگهانی، انعقاد پذیری بیش از حد خون (ترومبوفیلی)، پوکی استخوان، قوز و انحنای ستون فقرات (اسکولیوز) و انگشتان دست و پای بلند (آراکنوداکتیلی) اشاره نمود
هموسیستینوری
این بیماری، یک اختلال مرتبط با اسیدهای آمینه می باشد. افراد مبتلا به این بیماری برای هضم برخی از اسیدهای آمینه موجود در غذای روزانه خود، با مشکل مواجه می شوند. افراد مبتلا به این بیماری، توانایی هضم اسید آمینه متیونین را ندارند.
پروتئینها همراه با غذا وارد بدن ما می شوند، برای اینکه بدن بتواند از این پروتئینها استفاده کند، آنها باید به اجزا کوچکتری که اسید آمینه نام دارند، تبدیل شوند. سپس آنزیم ها، تغییراتی را در این اسیدهای آمینه ایجاد می کنند تا بدن بتواند از آنها استفاده کند. بیماری هموسیستینوری وقتی به وجود می آید که آنزیم ویژه ای که سیستاتیونین بتا سینتاز نامیده می شود یا به درستی عمل نکند و یا اصلا وجود نداشته باشد. وظیفه این آنزیم، هضم اسید آمینه متیونین است. در صورت فقدان آنزیم سیستاتیونین بتا سینتاز، سطح متیونین و یک اسید آمینه دیگر به نام هموسیستین در خون افزایش می یابد. افزایش سطح این مواد باعث ایجاد مشکلاتی در بدن می شود.
کودکان مبتلا به این بیماری در بدو تولد به نظر سالم می رسند. این بیماری در صورت عدم درمان، می تواند باعث تاخیر رشد و تاخیر یادگیری شود. این بیماری همچنین می تواند چشمها، استخوانها، قلب و عروق را نیز تحت تاثیر قرار دهد. دو نوع بیماری هموسیستینوری وجود دارد که نوع خفیف آن با ویتامین B6 قابل درمان است. علایم بیماری هموسیستینوری بسیار متنوع بوده و در افراد مختلف، متفاوت است.
رشد، یادگیری و رفتار:تاخیر در رشد و یادگیری معمولا بین ۳-۱ سالگی مورد توجه قرار می گیرد. مهمترین علایم، در کودکان درمان نشده شامل موارد زیر است:
عدم رشد مناسب
عدم وزن گیری مناسب
مشکلات رفتاری
تاخیر در خزیدن، راه رفتن و صحبت کردن
ناتوانی در یادگیری یا عقب ماندگی ذهنی
چشمها:اختلالات بینایی، معمولا یک سال پس از تولد بروز می کنند. در صورت عدم درمان، به تدریج شاهد در رفتگی عدسی چشم، افزایش فشار داخل چشم و بیماری آب سیاه و در ادامه نابینایی این بیماران خواهیم بود. در رفتگی عدسی چشم معمولا در کودکان ۸-۲ ساله ای که درمان نشده اند، دیده می شود.
استخوانها :نوجوانان و بالغین مبتلا به هموسیستینوری، اغلب بلند قامت و لاغر اندام هستند. این افراد ممکن است دستها، پاها و انگشتان بسیار بلندی داشته باشند. این بیماران معمولا بعد از سن ۱۰ سالگی، دچار استئوپروسیس می شوند. ضعف ماهیچه ها، به خصوص ماهیچه های پا، در برخی از مبتلایان دیده می شود.
قلب و عروق:هموسیستینوری در صورت عدم درمان، می تواند باعث انسداد عروق و در نتیجه مشکلات قلبی و سکته شود. در واقع مشکلات قلبی و سکته مهمترین عامل مرگ و میر در بیماران درمان نشده است.
دیگر عوارض بیماری هموسیستینوری :اغلب کودکان درمان نشده، دارای پوست و مو روشن هستند. در تعدادی از مبتلایان، التهاب پانکراس باعث ایجاد دردهای شدید می شود.
درمان هموسیستینوری
پزشک کودک شما با یک پزشک متخصص بیماریهای متابولیک و یک متخصص تغذیه همکاری خواهد کرد تا کودک شما را درمان کند. برای جلوگیری از عقب ماندگی ذهنی و مشکلات جسمی درمان فوری مورد نیاز است. اغلب مبتلایان، به رژیم غذایی فاقد متیونین، شیر خشک های خاص و دریافت برخی مکمل ها نیازمندند. شما به محض آگاهی از بیماری فرزندتان، باید معالجه را آغاز کنید. درمان معمولا مادام العمر است. در ادامه روش درمانی که به طور معمول برای بیماران هموسیستینوری پیشنهاد می شود، ذکر شده است:
رژیم غذایی با حداقل مقدار متیونین و غذاهای طبی:
این رژیم، شامل مواد غذایی با حداقل مقدار متیونین است. به عبارت دیگر کودک شما باید از شیر گاو، شیر خشک های معمولی، گوشت، تخم مرغ و پنیر پرهیز کند. همچنین آرد معمولی، باقلا خشک شده و مغزها (گردو و بادام و غیره) نیز دارای متیونین هستند و باید شدیدا محدود شوند. میوه ها و سبزیجات اغلب متیونین کمی دارند و در مقادیر تعیین شده، می توانند مورد استفاده قرار گیرند. همچنین مواد غذایی طبی وجود دارند که مناسب افراد هموسیستینوری می باشند همانند: آرد های مخصوص، پوره های مخصوص، نان و برخی از ماکارونی ها. پزشکان و متخصصین تغذیه رژیم غذایی مناسب کودک شما را به مرور زمان طراحی خواهند کرد. برنامه غذایی کودک شما به چندین پارامتر وابسته است همانند: سن کودک، وزن کودک، حال عمومی کودک و روند درمان کودک.
شیر خشک های مخصوص:
علاوه بر رژیم غذایی فاقد متیونین به برخی از بیماران، شیر خشک های خاص نیز داده می شود تا به جای شیر معمولی استفاده کنند. این شیر خشک ها، پروتئینها و مواد مغذی مورد نیاز کودک را تامین می کنند در حالیکه سطح متیونین و هموسیستین آنها در محدوده قابل قبول باقی می ماند. پزشکان به شما خواهند گفت که کدام شیر خشک و چه مقدار از آن برای کودک شما مناسب است.
مکمل ها
ویتامین B6برخی از کودکان مبتلا به هموسیستینوری، به درمان با ویتامین B6 پاسخ می دهند. در این کودکان، مصرف ویتامین B6 می تواند از مشکلات رفتاری و عقب ماندگی ذهنی جلوگیری کند. مصرف ویتامین B6 می تواند احتمال انسداد عروق و مشکلات بینایی و استخوانی را نیز در این بیماران کاهش دهد. از متخصصین، درباره مصرف این مکمل سوال کنید. ممکن است پزشک معالج از شما بخواهد که چند آزمایش خاص انجام دهید، تا او بتواند تاثیر این مکمل را در روند درمان کودک بررسی کند.
Betaineاین مکمل، یک ماده شبه ویتامین است که در برخی مواد غذایی وجود دارد و به شکل قرص نیز در دسترس می باشد. این دارو به کاهش سطح هموسیستین خون کمک می کند و می تواند یک مکمل مناسب برای بیمارانی باشد که به درمان با ویتامین B6 پاسخ نمی دهند. این مکمل می تواند، احتمال انسداد عروق را نیز کاهش دهد. پزشک معالج تصمیم خواهد گرفت که کودک شما به Betaine نیاز دارد یا خیر. هیچ دارویی را بدون مشورت با پزشک استفاده نکنید.
ویتامین B12سطح ویتامین B12 در بدن تعدادی از بیماران مبتلا به هموسیستینوری، پایین است. این افراد ممکن است، به تزریق ویتامین B12 نیاز داشته باشند. از پزشک معالج سوال کنید که آیا فرزند شما به ویتامین B12 نیاز دارد یا خیر.
اسید فولیکسطح اسید فولیک (نوعی ویتامین (B12 در بدن برخی از مبتلایان به هموسیستینوری پایین است. این بیماران ممکن است، به دریافت مکملهای اسید فولیک نیاز داشته باشند. اسید فولیک به کاهش سطح هموسیستین خون کمک می کند. درباره مصرف این مکمل با پزشک متخصص مشورت کنید.
L-Cystineسطح L-Cystine (یک نوع اسید آمینه) در بدن برخی از بیماران مبتلا به هموسیستینوری پایین است. این مکمل ممکن است در شیر خشکهای مخصوص بیماران هموسیستینوری وجود داشته باشد. در صورت عدم وجود این مکمل در رژیم غذایی، شما می توانید آن را به صورت خوراکی به فرزندتان بدهید. مصرف هر نوع دارو و مکمل باید با اجازه پزشک معالج باشد.
انجام تستهای منظم آزمایشگاهی
کودک شما به انجام تستهای مکرر خون و ادرار به منظور کنترل سطح اسیدهای آمینه نیاز خواهد داشت. نتیجه این تستهای آزمایشگاهی رژیم غذایی و داروهای کودک شما را تعیین خواهند کرد.
* احتمال انسداد عروق، سکته و مشکلات قلبی در زنان باردار مبتلا به هموسیستینوری بیشتر است. به همین دلیل در ماههای آخر بارداری به آنها، داروهای رقیق کننده خون داده می شود که مصرف این داروها باید تا ۶ هفته بعد از زایمان ادامه یابد. اجرای درمان معمولی هموسیستینوری به اضافه مصرف رقیق کننده های خونی، احتمال مرده زایی و سقط جنین را در زنان باردار کاهش می دهد.
اغلب کودکانی که بعد از تولد سریعا تحت درمان قرار گرفته اند، اکنون زندگی عادی همراه با رشد و نمو و سطح هوشی طبیعی دارند. درمان فوری احتمال انسداد عروق، سکته، مشکلات قلبی و مشکلات بینایی را در این بیماران، کاهش می دهد. هرچند در برخی از بیماران، علیرغم درمان باز هم در رفتگی عدسی چشم ایجاد می شود که با جراحی و یا دیگر متدها می توان آن را اصلاح کرد. کودکانی که درمان را دیر آغاز کرده اند، اغلب دارای مشکلات رفتاری و عقب ماندگی ذهنی هستند.
عوامل عدم فعالیت صحیح آنزیم
کنترل ساخت آنزیم ها در بدن به عهده ژنها است. افراد مبتلا به هموسیستینوری یک جفت ژن معیوب دارند که به درستی عمل نمی کنند. به خاطر این تغییر ایجاد شده در ژنها، یا آنزیم سیستاتیونین بتا سینتاز ساخته نمی شود و یا آنزیم ساخته شده به درستی فعالیت نمی کند.
وراثت هموسیستینوری
این بیماری در دختران و پسران به یک نسبت مشاهده می شود. هر فرد دارای دو ژن است که دستور ساخت آنزیم را میدهد. در کودکان مبتلا به هموسیستینوری هیچ یک از این دو ژن به درستی عمل نمی کنند. این کودکان هر یک از ژنهای معیوب را از یکی از والدین خود به ارث برده اند. به این نوع توارث، وراثت اتوزومال مغلوب می گویند. در والدین این کودکان، به ندرت اختلالی مشاهده می شود. اما هر یک از والدین یک تک ژن معیوب برای بیماری هموسیستینوری دارد. این والدین ناقل نامیده می شوند. ناقلین علامت بیماری را بروز نمی دهند زیرا دیگر ژن آنها سالم است. وقتی هر دو والدین ناقل باشند، ۲۵% احتمال تولد کودک کاملا سالم،۵۰% احتمال تولد کودک ناقل همچون والدین و ۲۵% احتمال تولد کودک مبتلا به هموسیستینوری در هر بارداری وجود دارد. مراکز مشاوره ژنتیک و مشاوران ژنتیک برای پاسخگویی به سوالات خانواده ها در دسترس می باشند. این مشاوران نحوه توارث بیماری را توضیح خواهند داد و همچنین راهنمای خانواده ها برای بارداری بعدی و مشاور آنها برای انجام تستهای تشخیصی برای دیگر اعضای خانواده خواهند بود.
آزمایش ژنتیک
امکان انجام تست ژنتیک وجود دارد. آزمایش ژنتیک که آزمایش DNA نیز نامیده می شود، تغییرات ایجاد شده در ژنها را بررسی می کند. در صورت هرگونه ابهام درباره آزمایش ژنتیک با پزشک متخصص بیماریهای متابولیک و یا با مشاور ژنتیک خود مشورت کنید. ممکن است، انجام آزمایش ژنتیک برای تشخیص بیماری ضروری نباشد، اما اگر امکان آن وجود داشته باشد، می تواند برای تشخیص قبل از تولد و یا تشخیص ناقلین مفید باشد.
آزمایشهای تاییدی
هموسیستینوری به وسیله تستهای خاصی که بر روی نمونه خون و ادرار انجام می شوند، شناسایی می گردد. در افراد مبتلا به این بیماری، معمولا سطح متیونین و هموسیستین خون افزایش می یابد. همچنین افزایش سطح هموسیستین در ادرار این بیماران مشاهده می شود. این بیماری به وسیله تستهای آنزیمی خاص، که بر روی نمونه پوست انجام می شود، تایید می گردد. درباره این تستها با مشاور ژنتیک و یا پزشک متخصص بیماریهای متابولیک مشورت کنید.
اگر یکی از فرزندان شما مبتلا به هموسیستینوری است، شما میتوانید آزمایش DNA را در طول بارداری بعدی انجام دهید. این آزمایش به شما خواهد گفت که، کودک بعدی شما مبتلا به این بیماری هست یا خیر. نمونه مورد نیاز برای انجام این تست از آمینو سنتز و یا CVS به دست می آید. اگر نتیجه آزمایش DNA قانع کننده نبود، تستهای آنزیمی دیگری نیز قابل انجام هستند. نمونه مورد نیاز برای این تستها از آمینوسنتز و یا CVS به دست می آید. والدین میتوانند تصمیم بگیرند که، تستهای آزمایشگاهی را در طول بارداری بر روی جنین انجام دهند و یا بعد از تولد کودک، تستها را روی نوزاد انجام دهند. مشاوران درباره این که کدام روش بهتر است به والدین توضیح خواهند داد.
امکان اینکه برادران و خواهران بزرگتر کودک مبتلا، که به طور طبیعی رشد کرده و به نظر سالم می رسند، به این بیماری مبتلا باشند کم است. هر چند جستجوی بیماری در دیگر اعضای خانواده بسیار مفید خواهد بود، زیرا از بسیاری از مشکلات جسمی پیشگیری خواهد کرد. از مشاور ژنتیک و یا پزشک متخصص سوال کنید که آیا دیگر اعضای خانواده باید تحت آزمایشهای تشخیصی قرار گیرند یا خیر. برادران و خواهران دیگر بیمار که هموسیستینوری ندارند ممکن است همانند والدین خود ناقل باشند. در ناقلین هیچ اختلالی مشاهده نمی شود. تستهای تشخیص ناقلین به جز در موارد خاص فقط در افراد بالای ۱۸ سال قابل انجام است. برادران و خواهران هریک از والدین نیز ۵۰% احتمال دارد که ناقل این بیماری باشند. اطلاع از ناقل بودن از این جهت که آنها نیز ممکن است بچه های مبتلا به این بیماری داشته باشند، حایز اهمیت است. هنگامی که هر دو والدین می دانند که آنها ناقل بیماری هستند و یا زمانی که یک کودک مبتلا به این بیماری در خانواده وجود دارد، دیگر کودکانی که در این خانواده ها متولد می شوند باید علاوه بر آزمایشهای غربالگری تحت آزمایشهای تشخیصی دقیقتر، برای هموسیستینوری قرار گیرند.
تستهای تشخیصی:
امکان انجام تستهای تشخیصی در دیگر افراد خانواده وجود دارد. نمونه مورد نیاز برای این تستها، نمونه خون و ادرار و پوست است.
تستهای تشخیص ناقلین:اگر یکی از افراد خانواده مبتلا به هموسیستینوری است، امکان انجام تست DNA در دیگر اعضای خانواده وجود دارد. اگر نتیجه این تست قانع کننده نبود، شما می توانید دیگر متدهای تشخیص ناقلین را انجام دهید. درباره این تستها با پزشک خود مشورت کنید.
شیوع بیماری هموسیستینوری چقدر است؟
شیوع این بیماری در ایالت متحده آمریکا در حدود یک مورد در هر۲۰۰۰۰۰ تولد تا یک مورد در هر ۳۰۰۰۰۰ تولد است. هموسیستینوری در تمامی نژادهای قومی سرتاسر جهان وجود دارد. هرچند پیش بینی می شود، که این بیماری در کشورهایی که ازدواجهای فامیلی رواج بیشتری دارد (همانند کشور ایران) شایعتر باشد.
این بیماری گاهی با نامهای زیر خوانده می شود:
هموسیستینمی
نقص سیستاتیونین بتا سینتاز
نقص CBS
.
در خصوص سایر زیر گروه های اختلالات اسید های آمینه مواردزیر وجود دارند که در بخش های اختصاصی آنها نیز مورد بحث و تبادل نظر قرار گرفته اند:
- متابولیسم هیستیدین، تریپتوفان و لایزین
- متابولیسم سرین، گلیسین و گلیسرات
- متابولیسم اورنیتین یا پرولین
- اختلال در نقل و انتقال اسید های آمینه در سلول
- متابولیسم اسید آمینه
- چرخه گاما گلوتامیل
- متابولیسم سایر پپتید ها
اختلالات متابولیسم کربوهیدرات ها
اختلالات مادرزادی کربوهیدرات ها به زیر گروه های مختلفی تقسیم می شود که در ادامه به صورت مختصر به هر یک از آنها پرداخته شده است.
گالاکتوزومی کلاسیک
گالاکتوزومی (Galactosomia) یک بیماری اتوزومی مغلوب می باشد که به دلیل نقص در آنزیم گالاکتوز 1- فسفات یوریدیل ترانسفراز ایجاد می شود. این آنزیم برای متابولیسم و هضم قند گالاکتوز خوراکی مورد نیاز بدن می باشد. افرادی که در ژن مسئول ساخت این آنزیم دارای نقص باشند مقادیر افزایش یافته ای از گالاکتوز در خون و مایعات بدن آنها ایجاد می شود. نوزادان مبتلا علائمی همانند استفراغ، رخوت، نارسایی رشد، و زردی را در طی هفته دوم بعد از تولد نشان می دهند. اگر درمان صورت نگیرد در نهایت بیماری منجر به عقب ماندگی ذهنی، کاتاراکت (آب مروارید) و سیروز کبدی خواهد شد. برای درمان این نوزادان ضروری است تا از شیر های فاقد لاکتوز استفاده شود. لاکتوز یک قند دو واحدی است که پس از تجزیه در بدن به یک گلوکوز و یک گالاکتوز تجزیه می شود. وجود گالاکتوزمی را می توان در غربالگری نوزادان تشخیص داد.
عدم تحمل فرکتوز ارثی
عدم تحمل فروکتوز ارثی یک بیماری اتوزومی مغلوب است که که در اثر نقص آنزیم فروکتوز 1- فسفات آلدولاز ایجاد می شود. قند فروکتوز در موادی همانند عسل، میوه و سبزیجات به وفور یافت می شود و همچنین این قند در ترکیب ساکاروز یا همان شکر خوراکی (به صورت ترکیب دو تایی با گلوکوز) دیده می شود. افراد مبتلا در سنین مختلف و بسته به اینکه این قند از چه زمانی به رژیم غذایی آنها وارد می شود شناسایی می شوند. علائم معمولا خفیف است ولی می تواند در مواردی شدت بیشتری هم نشان دهد و شامل استفراغ، زردی و تشنج می باشد. با کنترل مقادیر فرکتوز ورودی در غذای بیمار می توان بیماری را به خوبی مدیریت کرد.
اختلالات ذخیره گلیکوژن
گلیکوژن شکل ذخیره ای قند گلوکز در عضلات و کبد می باشد که به صورت یک پلیمر (زنجیره) از مولکول های گلوکز به وجود می آید. این ماده در زمان نیاز بدن تجزیه شده و به قند گلوکز تبدیل می شود و مورد استفاده عضلات و سایر بافت ها قرار می گیرد. چندین آنزیم مختلف در ساخت و تجزیه گلیکوژن نقش دارند که اختلال در هر یک از آنها می تواند به یکی از انواع بیماری های ذخیره گلیکوژن (GSD یا Glycogen Storage Disorder) منجر شود. در مجموع حدود 30 نوع از بیماری ذخیره گلیکوژن شناخته شده است که مهمترین آنها عبارتند از:
- بیماری ون ژیرکه (Von Gierke)
- بیماری پمپه (Pompe Disease)
- بیماری کوری (Cori Disease)
- بیماری مک آردل (McArdle Disease)
- نقص فسفوریلاز کبدی
سایر اختلالات متابولیسم کربوهیدرات
- متابولیسم پنتوز ها
- متابولیسم گلیسرول
- متابولیسم گلی اکزالات
- انتقال گلوکز
- گلوکونئوژنز
اختلالات اکسیداسیون اسیدهای چرب (FAOD) گروهی از بیماری های نادر ارثی هستند که توسط آنزیم هایی که به درستی کار نمی کنند ایجاد می شوند. تعدادی آنزیم برای تجزیه چربی ها در بدن مورد نیاز است (فرآیندی به نام اکسیداسیون اسیدهای چرب). مشکلات مربوط به هر یک از این آنزیم ها می تواند باعث اختلال اکسیداسیون اسیدهای چرب شود. افراد مبتلا به FAOD نمی توانند به درستی چربی را از طریق غذایی که می خورند یا از چربی ذخیره شده در بدنشان تجزیه کنند. بدن سالم از گلوکز (قند) برای انرژی استفاده می کند. وقتی بدن از تمام گلوکز موجود استفاده می کند، از چربی انرژی می گیرد. نوزاد مبتلا به اختلال اکسیداسیون اسیدهای چرب نمی تواند از چربی برای انرژی استفاده کند. این می تواند باعث کاهش قند خون و مواد مضر در خون او شود.
FAOD ها به صورت اتوزومی مغلوب به ارث می رسند و هم در مردان و هم در زنان تأثیر می گذارند. این بدان معناست که آنها از طریق ژن ها از والدین به فرزند منتقل می شوند. ژن ها بخش هایی از سلول های بدن شما هستند که دستورالعمل هایی را برای نحوه رشد و عملکرد بدن ذخیره می کنند.
علائم و درمان بین FAOD های مختلف متفاوت است. آنها همچنین می توانند از فردی به فرد دیگر با همان FAOD متفاوت باشند. برگه های اطلاعاتی برای هر FAOD خاص را ببینید.
چه اختلالات اکسیداسیون اسیدهای چرب در غربالگری نوزادان آزمایش می شود؟
نقص جذب کارنیتین (CUD)
کودکان مبتلا به CUD معمولاً بین تولد تا هفت سالگی علائم دارند، اما برخی از آنها هرگز علائمی ندارند. در صورت عدم درمان، CUD می تواند باعث آسیب مغزی یا مرگ شود. CUD هر سال از هر 100000 نوزاد در ایالات متحده یک نوزاد را تحت تاثیر قرار می دهد. با این حال، در جمعیت های خاصی شایع تر است. تخمین زده می شود که از هر 40000 نوزادی که در ژاپن به دنیا می آیند، یک نوزاد را تحت تاثیر قرار می دهد.
کمبود هیدروکسی سیسیل-COA دهیدروژناز با زنجیره بلند (LCHAD)
از هر 75000 نوزاد هر سال یک نوزاد با LCHAD در ایالات متحده متولد می شود. کودکان معمولاً اندکی پس از تولد علائم دارند. LCHAD ممکن است در افراد فنلاند شایع تر باشد.
کمبود ACYL-COA دهیدروژناز با زنجیره متوسط (MCAD)
MCAD از هر 15000 نوزاد در ایالات متحده یک نفر را تحت تاثیر قرار می دهد. علائم اغلب در نوزادان یا کودکان خردسال رخ می دهد و شامل هیپوگلیسمی (قند خون پایین)، بیماری کبد یا مرگ ناگهانی است. در افراد با اصل و نسب اروپای شمالی بیشتر دیده می شود. در صورت عدم درمان، MCAD می تواند باعث آسیب مغزی و مشکلات تنفسی شود.
کمبود پروتئین سه عملکردیTrifunctional Protein Deficiency
TFP یک بیماری بسیار نادر است و کمتر از یک نوزاد از هر 100000 نوزادی که هر سال با TFP در ایالات متحده متولد می شود را تحت تاثیر قرار می دهد. سه نوع TFP وجود دارد:
نوزادان اولیه TFP می توانند علائم و نشانه ها را در هر زمانی از سن 2 سالگی داشته باشند. اگر درمان نشود، این نوزادان اغلب در سن 3 سالگی به دلیل مشکلات قلبی یا ریوی می میرند.
کودکان TFP دوران کودکی پس از یک سالگی علائم و نشانه هایی دارند. تکرار علائم TFP در کودکان می تواند باعث آسیب مغزی شود و منجر به مشکلات یادگیری یا ناتوانی ذهنی شود. برخی از کودکان مبتلا به TFP در دوران کودکی ممکن است پس از ورزش سخت، بیماری یا نوعی استرس بدنی دیگر احساس ضعف یا درد کنند.
TFP خفیف - این نوع TPF نادر است. علائم هر زمان بعد از 2 سالگی شروع می شود، از جمله در بزرگسالی.
Very-Long-Chain ACYL-COA Dehydrogenase Deficiency (VLCAD)
در صورت عدم درمان، VLCAD می تواند باعث آسیب مغزی و حتی مرگ شود. بیش از یک نوزاد از هر 75000 نوزاد هر سال با VLCAD در ایالات متحده متولد می شود. سه نوع VLCAD وجود دارد:
نوزاد VLCAD اولیه تا 4 ماهگی علائم دارد. حدود نیمی از نوزادان مبتلا به VLCAD زودرس VLCAD دارند.
علائم VLCAD دوران کودکی نزدیک به اولین تولد نوزاد یا در اوایل کودکی ظاهر می شود. حدود یک سوم افراد مبتلا به VLCAD در دوران کودکی VLCAD دارند.
VLCAD بزرگسالان - حدود 20 درصد از VLCAD دارای این نوع هستند. علائم در سنین نوجوانی یا بزرگسالی ظاهر می شود.
Carnitine Palmitoyltransferase I Deficiency (CPT-1A)
CPT-1A یک بیماری نادر است. کمتر از 50 نفر مبتلا به CPT-1A گزارش شده است. این بیماری در گروههای قومی خاص، از جمله در جمعیت هوتریت در آمریکای شمالی و در مردم اینوئیت آلاسکا و کانادا شایعتر است. CPT-1A در یک نوزاد از هر 1200 نوزاد متولد شده در جمعیت هوتریت آمریکا یافت می شود.
Carnitine Palmitoyltransferase II Deficiency (CPT-II)
CPT-II یک بیماری نادر است. سه شکل اصلی CPT-II وجود دارد که در سن شروع آنها متفاوت است. شکل نوزادی در 18 مورد گزارش شده است. شکلی که نوزادان و کودکان خردسال را تحت تاثیر قرار می دهد حداقل در 30 مورد توصیف شده است. کلاسیک CPT-II عموماً بزرگسالان را تحت تأثیر قرار می دهد و شایع ترین شکل آن است. در بیش از 300 مورد گزارش شده است. بروز کلی CPT-II ناشناخته است.
Short-Chain ACYL-COA Dehydrogenase Deficiency (SCAD)
تخمین زده می شود که SCAD از هر 40000 تا 100000 نوزاد یک نفر را تحت تاثیر قرار دهد. اکثر افرادی که به عنوان مبتلا به SCAD شناخته می شوند هرگز علائمی را تجربه نمی کنند.
Glutaric Acidemia, Type II (GA-2)
GA-2 یک بیماری بسیار نادر است و تعداد دقیق افراد مبتلا به GA-2 در حال حاضر ناشناخته است. GA-2 می تواند باعث هیپوگلیسمی، تون عضلات ضعیف، مشکلات قلبی شدید و مرگ شود. GA-2 یک وضعیت متفاوت از GA نوع 1 است.
علائم و نشانه های این اختلالات چیست؟
علائم و نشانه های این اختلالات معمولا در نوزادان یا کودکان خردسال بروز می کند. برخی از این اختلالات می توانند ابتدا در بزرگسالان ظاهر شوند، اما این نادر است. بیماری یا عفونت، خوردن غذاهای نادرست، یا ماندن برای مدت طولانی بدون غذا خوردن می تواند علائم و نشانه های این اختلالات را ایجاد کند. علائم و نشانه ها عبارتند از:
تغییرات در رفتار
اسهال، حالت تهوع و استفراغ
خواب آلودگی
تب
سر و صدا
اشتهای کم
از دست دادن احساس در بازوها و پاها
قند خون پایین
درد عضلانی، گرفتگی یا ضعف
مشکلات بینایی
این اختلالات چه مشکلاتی برای سلامتی ایجاد می کنند؟
در صورت عدم درمان، برخی از این اختلالات می توانند باعث مشکلات سلامتی شوند، از جمله:
ضربه مغزی
مشکلات تنفسی
کما
مشکلات قلبی، کبدی و ریوی
ناتوانی های ذهنی و رشدی
تشنج
در صورت عدم درمان، برخی از اختلالات می توانند منجر به مرگ شوند.
درمان هایی که اغلب برای کودکان مبتلا به اختلالات اکسیداسیون اسیدهای چرب توصیه می شود عبارتند از:
پرهیز از روزه گرفتن
درمانهای رژیمی - ممکن است کودک شما نیاز به رژیم غذایی محدود داشته باشد تا از غذاهای خاصی که بدنش نمیتواند تجزیه کند اجتناب کند. یک متخصص تغذیه یا یک متخصص تغذیه می تواند به شما در برنامه ریزی یک رژیم غذایی سالم برای فرزندتان کمک کند.
مکمل ها و داروهایی مانند ال کارنیتین، روغن MCT، ریبوفلاوین، گلیسین، DHA

ارسال به دوستان